Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Artificial gene synthesis wikipedia , lookup
Protein–protein interaction wikipedia , lookup
Peptide synthesis wikipedia , lookup
Point mutation wikipedia , lookup
Amino acid synthesis wikipedia , lookup
Biosynthesis wikipedia , lookup
Ribosomally synthesized and post-translationally modified peptides wikipedia , lookup
Two-hybrid screening wikipedia , lookup
Ancestral sequence reconstruction wikipedia , lookup
Biochemistry wikipedia , lookup
Genetic code wikipedia , lookup
Sequence Based Analysis Tutorial
NIH Proteomics Workshop
Lai-Su Yeh, Ph.D.
Protein Information Resource at
Georgetown University Medical Center
Retrieval, Sequence Search &
Classification Methods
Retrieve protein info by text / UID
Sequence Similarity Search
BLAST, FASTA, Dynamic Programming
Family Classification
Patterns, Profiles, Hidden Markov Models,
Sequence Alignments, Neural Networks
Integrated Search and Classification System
2
Sequence Similarity Search (I)
Based on Pair-Wise Comparisons
Dynamic Programming Algorithms
Global Similarity: Needleman-Wunch
Local Similarity: Smith-Waterman
Heuristic Algorithms
FASTA: Based on K-Tuples (2-Amino Acid)
BLAST: Triples of Conserved Amino Acids
Gapped-BLAST: Allow Gaps in Segment Pairs
PHI-BLAST: Pattern-Hit Initiated Search
PSI-BLAST: Position-Specific Iterated Search
3
Sequence Similarity Search (II)
Similarity Search Parameters
Scoring Matrices – Based on Conserved Amino
Acid Substitution
Dayhoff Mutation Matrix, e.g., PAM250 (~20%
Identity)
Henikoff Matrix from Ungapped Alignments,
e.g., BLOSUM 62
Gap Penalty
Search Time Comparisons
Smith-Waterman: 10 Min
FASTA: 2 Min
BLAST: 20 Sec
4
Feature Representation
Features of Amino Acids: Physicochemical Properties,
Context (Local & Global) Features, Evolutionary Features
Alternative Amino Acids: Classification of Amino Acids To
Capture Different Features of Amino Acid Residues
Alphabet
AA Identity
Exchange Group
Charge/Polarity
Hydrophobicity
Structural
2D Propensity
Size
20
6
4
3
3
3
Features
Sequence Identity
EvolutionSubstitution
Charge and Polarity
Hydrophobicity
Surface Exposure
Secondary Structure
Membership
A,C,D,E,F,G,H,I,K,L,M,N,P,Q,R,S,T,V,W,Y
{HRK}{DENQ}{C}{STPAG}{MILV}{FYW}
{HRK} {DE} {CTSGNQY} {PMLIVFW}
{DENQRK} {CSTPGHY} {AMILVFW}
{DENQHRK} {CSTPAGWY} {MILVF}
{AEQHKMLR} {CTIVFYW} {SGPDN}
5
Substitution Matrix
Likelihood of One Amino Acid Mutated into Another Over
Evolutionary Time
Negative Score: Unlikely to Happen (e.g., Gly/Trp, -7)
Positive Score: Conservative Substitution (e.g., Lys/Arg, +3)
High Score for Identical Matches: Rare Amino Acids (e.g., Trp, Cys)
6
Secondary Structure Features
a Helix Patterns of Hydrophobic Residue Conservation Showing I,
I+3, I+4, I+7 Pattern Are Highly Indicative of an a Helix
(Amphipathic)
b Strands That Are Half Buried in the Protein Core Will Tend to Have
Hydrophobic Residues at Positions I, I+2, I+4, I+6
7
BLAST
BLAST (Basic Local Alignment Search Tool)
Extremely fast
Robust
Most frequently used
It finds very short segment pairs (“seeds”) between the query
and the database sequence
These seeds are then extended in both directions until the
maximum possible score for extensions of this particular
seed is reached
8
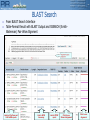
BLAST Search
From BLAST Search Interface
Table-Format Result with BLAST Output and SSEARCH (SmithWaterman) Pair-Wise Alignment
Links to iProClass and
UniProtKB reports
Link to NCBI
taxonomy
Link to PIRSF
report
Click to see
SSearch alignment
Click 9
to see
alignment
Blast Result & Pairwise Alignment
BLAST
Aligment
10
How do you build a tree?
Pick sequences to align
Align them
Verify the alignment
Keep the parts that are aligned correctly
Build and evaluate a phylogenetic tree
Integrated Analysis
11
Multiple Sequence Alignment: CLUSTALW
Pairwise alignment:
Calculate distance matrix
Mean number of
differences per residue
Unrooted Neighbor-Joining Tree
Branch length drawn
to scale
Rooted NJ Tree
(guide tree)
Root place at a position where
the means of the branch lengths
on either side of the root are
equal
Progressive
Alignment guided
by the tree
Alignment starts from the tips of
the tree towards the root
Thompson et al., NAR 22, 4675 (1994).
12
PIR Multiple Alignment and Tree
From Text/Sequence Search Result or CLUSTAL W Alignment
Interface
13
14
PIR Pattern Search
Signature Patterns for Functional Motifs
From Text/Sequence Search Result or Pattern Search Interface
Alignment of a region involved
in catalytic activity
A
P-[IV]-[WY]-x(3)-H-[MR]-V-x(3,4)-Q-x(1,2)-D-x(4,5)-G-A-N
Create Pattern and search in database:
P-[IV]-[WY]-x(3)-H-[MR]-V-x(3,4)-Q-x(1,2)-D-x(4,5)-G-A-N
Test sequence against PROSITE database
B
O05689
15
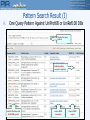
Pattern Search Result (I)
A.
One Query Pattern Against UniProtKB or UniRef100 DBs
Display the query
pattern
Indicate pattern
sequence region(s)
Links to iProClass and
UniProtKB reports
Link to NCBI
taxonomy
Link to PIRSF
report
16
Pattern Search Result (II)
B.
One Query Sequence Against PROSITE Pattern Database
17
Profile Method
Profile: A Table of Scores to Express Family Consensus Derived
from Multiple Sequence Alignments
Num of Rows = Num of Aligned Positions
Each row contains a score for the alignment with each
possible residue.
Profile Searching
Summation of Scores for Each Amino Acid Residue along
Query Sequence
Higher Match Values at Conserved Positions
18
Prosite PS50157 profile for Zinc finger C2H2
19
1
PIRSF scan
Search One Query
Protein Against all
the Full-length and
Domain HMM models
for the fully curated
PIRSFs by HMMER
The matched regions
and statistics will be
displayed.
Shows PIRSF that the
query belongs to
Statistical data for all
domains
Statistical data
per domain
Alignment
with
consensus
sequence
20
Lab Section
21
Rat eye lens phosphoproteomics in normal and cataract
Kamei et al., Biol. Pharm. Bull., 2005.
Normal
pI
(+)
More phosphorylated spots
in cataract sample.
Digestion and MS from Spot
16 gave these peptides:
Mw
(-)
Cataract
MDVTIQHPWFKR
ALGPFYPSR
CSLSADGMLTFSG
YRLPSNVDQSALS
We want to identify the protein(s) that contain these peptides
Use Peptide Search
22
Peptide Search
23
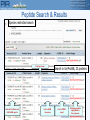
Peptide Search & Results
Species restricted search
Sorting arrows
Links to iProClass and
UniProtKB reports
Link to NCBI
taxonomy
Search in UniProtKB, 23 proteins
Link to PIRSF
report
Matching peptide
highlighted in
the sequence
24
Batch Retrieval Results (I)
• Retrieve multiple proteins in from
iProClass using a specific identifier or
a combination of them
• Provides a means to easily retrieve
and analyze proteins when the
identifiers come from different
databases
Retrieve more
sequences
25
ID Mapping
26
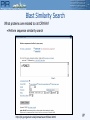
Blast Similarity Search
What proteins are related to rat CRYAA?
• Perform sequence similarity search
>P24623
http://pir.georgetown.edu/pirwww/search/blast.shtml
27
Pairwise Alignment
29
PIR Text Search
(http://pir.georgetown.edu/search/textsearch.shtml)
UniProtKBDatabase
and unique UniParc
sequences
Let’s search for
human crystallins
PIR protein family
classification
database
30
Let’s look for crystallins which have 3D structure
Refine your
search or start
over
Display PDB ID
31
Domain Display allows to compare simultaneously Pfam domains present in
multiple proteins
Share same domain
architecture
Let’s perform a multiple alignment on the sequences containing PF00030
32
Multiple Alignment
33
Interactive Phylogenetic Tree and Alignment
Beta B1 and gamma crystallins share the same domains, SCOP fold
and share significant sequence similarity suggesting that they are
34
related
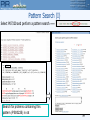
Pattern Search (I)
Select P07320 and perform a pattern search
Search for proteins containing this
pattern (PS00225) in rat
35
Pattern Search Result
Beta and gamma
Crystallins have
multiple copies of
this pattern
36