Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
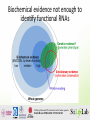
RNA-‐seq Introduc1on Promises and pi7alls RNA gives informa1on on which genes that are expressed How DNA get transcribed to RNA (and some1mes then translated to proteins) varies between e. g. -‐Tissues -‐ Cell types -‐ Cell states -‐Individuals -‐Cells RNA gives informa1on on which genes that are expressed How DNA get transcribed to RNA (and some1mes then translated to proteins) varies between e. g. -‐Tissues -‐ Cell types -‐ Cell states -‐Individuals RNA gives informa1on on which genes that are expressed How DNA get transcribed to RNA (and some1mes then translated to proteins) varies between e. g. -‐Tissues -‐ Cell types -‐ Cell states -‐Individuals RNA flavors (pre sequencing era) • House keeping RNAs – rRNAs, tRNAs, snoRNAs, snRNAs, SRP RNAs, cataly1c RNAs (RNAse E) • Protein coding RNAs – (1 coding gene ~ 1 mRNA) • Regulatory RNAs – Few rare examples ENCODE, the Encyclopedia of DNA Elements, is a project funded by the Na1onal Human Genome Research Ins1tute to iden1fy all regions of transcrip1on, transcrip1on factor associa1on, chroma1n structure and histone modifica1on in the human genome sequence. ENCyclopedia Of Dna Elements Different kind of RNAs have different expression values Landscape of transcrip/on in human cells, S Djebali et al. Nature 2012 What defines RNA depends on how you look at it Coverage Variants Abundance House keeping RNAs mRNAs Regulatory RNAs Novel intergenic None Adapted from Landscape of transcrip/on in human cells, S Djebali et al. Nature 2012 Defining func1onal DNA elements in the human genome • Statement • Consequence – A priori, we should not expect the – Thus, one should have high transcriptome to consist exclusively of func1onal RNAs. • Why is that – Zero tolerance for errant transcripts would come at high cost in the proofreading machinery needed to perfectly gate RNA polymerase and splicing ac1vi1es, or to instantly eliminate spurious transcripts. – In general, sequences encoding RNAs transcribed by noisy transcrip1onal machinery are expected to be less constrained, which is consistent with data shown here for very low abundance RNA confidence that the subset of the genome with large signals for RNA or chroma1n signatures coupled with strong conserva1on is func1onal and will be supported by appropriate gene1c tests. – In contrast, the larger propor1on of genome with reproducible but low biochemical signal strength and less evolu1onary conserva1on is challenging to parse between specific func1ons and biological noise. This is of course not without an debate Variants Abundance Most ‘‘Dark Matter’’ Transcripts Are Associated With Known Genes Perspective Harm van Bakel1, Corey Nislow1,2, Benjamin J. Blencowe1,2, Timothy R. Hughes1,2* 1 Banting and Best Department of Medical Research, University of Toronto, Toronto, Ontario, Canada, 2 Department of Molecular Genetics, University of Toronto, Toronto, Ontario, Canada The Reality of Pervasive Transcription Abstract Michael B. Clark1, Paulo P. Amaral1., Felix J. Schlesinger2., Marcel E. Dinger1, Ryan J. Taft1, John L. Perspective 4 5 6 portion of the mammalian 8 A series of3,reports the last few yearsF.have indicated that aV.much larger is Chris over P. Ponting , Peter Stadler , Kevin Morris , Antonin Morillon7, Joelgenome S. Rozowsky , Rinn transcribed than can be accounted for by currently and nature of10these additional 8 9 annotated genes, but the quantity 10 Mark B. Gerstein , Claes Wahlestedt , Yoshihide Hayashizaki , Piero Carninci , Thomas R. Gingeras2*, transcripts remains unclear. Here, we have used data from single- and paired-end RNA-Seq and tiling arrays to assess the John Mattick1*of transcripts in PolyA+ RNA from human and mouse tissues. Relative to tiling arrays, RNA-Seq quantity andS.composition Response to ‘‘The Reality of Pervasive Transcription’’ identifies many transcribed regions of(‘‘seqfrags’’) outsideQueensland, known exons and2 Watson ncRNAs. Most nonexonic seqfrags are inHarbor Laboratory, 1 Institute for fewer Molecular Bioscience, University Queensland, Brisbane, Australia, School of Biological Sciences, Cold Spring 1 1,2 pre-mRNAs. The chromosomal 1,2 locations of the majority 1,2of introns, the possibility that States they areAmerica, fragments Harm Bakel , Corey Nislow , Benjamin Blencowe , Timothy R. Hughes * Genomics Unit, Coldraising Spring Harbor, Newvan York, United of 3 Broadof Institute, Cambridge,J.Massachusetts, United States of America, 4 MRC Functional intergenic seqfrags in RNA-Seq data are near known genes, consistent with alternative cleavage and polyadenylation siteof Leipzig, Leipzig, Department of 1 Physiology, Anatomy and Genetics, University of Oxford, Kingdom, Department of Computer Science, University Banting and Best Department of Medical Research and Oxford, TerrenceUnited Donnelly Centre5for Cellular and Biomolecular Research, University of Toronto, Toronto, Ontario, usage,Germany, promoterand terminator-associated transcripts, or newResearch alternative exons; indeed, reads bridge splice sites Curie, 6 Department Molecular and Experimental Medicine, Scripps Institute, La Jolla, California, Unitedthat States of America, 7 Institut UMR3244Canada, 2ofDepartment of Molecular Genetics, University of Toronto, Toronto, Ontario, Canada identified 4,544 new exons,Paris, affecting genes. Biology Most of remaining to eitherUnited singleStates reads that 9 University of Pavillon Trouillet Rossignol, France, 83,554 Computational andthe Bioinformatics, Yaleseqfrags University,correspond New Haven, Connecticut, of America, display characteristics random sampling from low-level background or Institute, severalTsurumi-ku, thousandYokohama, small transcripts (median Miami, Miami, Florida, of United States of America, 10 OmicsaScience Center, RIKEN Yokohama Japan tic’’ transcriptsconservation greatly increases their Kanagawa, emphasized the lack of abundant pervasive Clark al. criticize aspects length = 111 bp) present atethigher levels, several which also tendofto display sequence and originate from regions with [7,8].transcripts, their number andtranscription our conclude study [1],that, andwhile specifically challenge open chromatin. We there are bona fide abundance new intergenic abundance in is our study. Clark et al. cite papers that have previously documented that transcribed the phrase quoted our assertiontothat the degree pervasive generally low in comparison known exons, of and the genome isWe notacknowledge as pervasively as previously reported. pervasive transcription, and point out that by Clark et al. in our Author Summary transcription has previously been overstatribosomal portion polyA+approaches transcrip- have been Current estimates indicate that only Previous Evidence for Pervasive severalof the different should have read ‘‘stably transcribed’’, or ed. We disagree with much of their Citation: van Bakel H, Nislow C, Blencowe BJ, Hughes TR (2010) Most ‘‘Dark Matter’’ Transcripts Are Associated With Known Genes. PLoS Biol 8(5): e1000371. tome [7,8,10,14–19], normalization ap- that Clark about 1.2% of the mammalian genome some equivalent, rather than simply ‘‘tranused as confirmation. We believe reasoning and their interpretationTranscription of our doi:10.1371/journal.pbio.1000371 proaches were used to reduce thewhat quantity codes for amino acids in proteins. Howscribed’’. But this does not change the fact et al. misinterpret can be claimed work. For example, many of our concluAcademic Editor: Sean R. Eddy, HHMI Janelia Farm, United States of America highly expressed transcripts in these The conclusion the disagree mammalian ever, mounting evidence over the past that wethat strongly with theoffundafrom much of the literature in this area, and sions are based on overall sequence read Received December 3, 2009; Accepted April 9, 2010; Published May 18, 2010 and are implicit genome transcribed (i.e., bycDNA decade has suggested that while the vast majormental argument put forward Clarkanalyses fail [7,8], to acknowledge known inweaknesses in distributions, Clark et al. focus on is pervasively Copyright: Bakel et al.isThis is an open-access distributed under the Commons Attribution License, which permits tiling array approaches. This studies. was neces‘‘that the majority ofwhich its ofbases are associity !of2010 thevangenome transcribed, well article(sets et the al., terms is Creative that the genomic area some of these We previously transcript units and seqfrags of unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. sary imto allowreviewed the detection rarer [9]. (oftenFor example, with at corresponding least one primary transcript’’ is more beyond the boundaries known A genes, to transcripts theseofissues overlappingof reads). key apoint ated is that Funding:phenomenon This work was supported byasGenome Canadatran(http://www.genomecanada.ca) through the Ontario Genomics Ontario Research Fund, and cellthe type–restricted [1,13,16,19,20]) tran- detected in [1]) the was based onthan multiple lines abundance. ofInstitute, known pervasive portant their relative This the number of transfrags one can derive a robust March of Dimes (http://www.marchofdimes.com). HvB wasestimate supportedof by the Netherlands Organization for Scientific Research (NWO; http://www.nwo.nl) (grant scripts. evidence. Both large-scale cDNA sequencscription [1].Canadian Challenging this an (CIHR; viewpoint makes sense toThe us. funders Given the permuted tiling array data can be as high no. 825.06.033) and the Institutes of Health Research http://www.cihr-irsc.gc.ca/) (grantlittle no. 193588). had no role in study design, relative amounts ofview, different transcript The evidence and hybridization to genome-wide data collection analysis, decision to publish, or by preparation the manuscript. articleandpublished inwithout PLoS Biology van of ing various sources of extraneous sequence as itfor is pervasive in the realtranscription data [10]. In addition, a types having a complete reconalso includes observations a wide tiling arrays were the major empirical Bakel et al. concluded that ‘‘the genome is reads, both biological and laboratorycommon form from of ‘‘validation’’ in these Competing Interests: The authors have declared that no competing interests exist. struction of every single transcript. variety independent sources of data. Analysis of full-length (see below), it isanalysis expected that with of other not as APA, pervasively as polyadenylation; previouspapers RT-PCRtechniques or RACE, but these In transcribed this brief and response, we firstBW,revisit Abbreviations: alternative cleavage bandwidth derived parameter; CAGE, capped of gene expression; CNV, copyisnumber reviews [21] [22] references). cDNAs from many tissues and pasRNA, developsufficient sequencing depth the(see entire reported’’ [2] and that the oftranscription, variation; ly lincRNAs, large intervening noncoding RNAs; ncRNAs, noncoding RNAs; ORF, open reading frames; promoter-associated RNA; TSS, and transcription approaches arefor generally semi-quantitative what is meant by majority pervasive Biochemical evidence not enough to iden1fy func1onal RNAs Defining functional DNA elements in the human genome Kellis M et al. PNAS 2014;111:6131-6138 One gene many different mRNAs RNA-seq: alternative splicing • RNA seq course The RNA seq course • • • • • • • From RNA seq to reads Mapping reads programs Transcriptome reconstruc1on using reference Transcriptome reconstruc1on without reference QC analysis sRNA analysis Differen1al expression analysis – mRNAs – miRNAs • Genome annota1on using RNA and other sources • Differen1al expression using mul1 variate analysis • RNA long read analysis From RNA to short reads Promises and pi7alls Long reads • • • • • • Low throughput (-‐) Complete transcripts (+) Only highly expressed genes (-‐-‐) Expensive (-‐) Low background noise (+) Easy downstream analysis (+) • • • • • • • High throughput (+) Only known sequences (-‐) Limited dynamic range (-‐) Cheap (+) High background noise (-‐) Not strand specific (-‐) Well established downstream methods (+) RNAseq • • • • • • • • High throughput Frac1ons of transcripts Full dynamic range Unlimited dynamic range Cheap Low background noise Strand specificity Re-‐sequencing 10000 1000 Signal Micro Arrays EST 100 MicroArray 10 RNAseq 1 1 10 100 1000 10000 # trancripts/cell 100000 1000000 (+) (-‐) (+-‐) (+) (+) (+) (+) (+) How are RNA-‐seq data generated? Sampling process RNA seq reads correspond directly to abundance of RNAs in the sample RNA to reads RNA-‐> enrichments -‐> AAAAAAAA PolyA (mRNA) RiboMinus (-‐ rRNA) Size <50 nt (miRNA ) ….. Size of fragment Strand specific 5’ end specific 3’ end specific ….. library -‐> reads -‐> Single end (1 read per fragment) Paired end (2 reads per fragment) Transcriptome assembly using reference Transcriptome assembly without reference Mapping long reads to reference Quality control -‐samples might not be what you think they are • Experiments go wrong – 30 samples with 5 steps from samples to reads has 150 poten1al steps for errors – Error rate 1/100 with 5 steps suggest that one of every 20 samples the reads does not represent the sample • Mixing samples – 30 samples with 5 steps from samples to reads has ~24M poten1al mix ups of samples – Error rate 1/ 100 with 5 steps suggest that one of every 20 sample is mislabeled • Combine the two steps and approximately one of every 10 samples are wrong RNA QC Read quality Mapping sta1s1cs Transcript quality Compare between samples Differen1al expression analysis using univariate analysis Typically univariate analysis (one gene at a 1me) – even though we know that genes are not independent Gene set analysis and data integra1on cleaved by RNase Y of total virulence factors are affected by RNase Y deletion (88% downregulated) SCP hidden by gene downregulation ? microRNA analysis (Jakub) (Berezikov et al. Genome Research, 2011.) Single cell RNA-‐seq analysis (Sandberg, Nature Methods 2014) Long reads Short reads Long reads Short reads Long reads