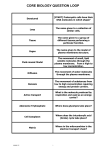
Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
SINGLE MOLECULE OPTICAL MAGNETIC TWEEZERS MICROSCOPY STUDIES OF PROTEIN DYNAMICS Qing Guo A Dissertation Submitted to the Graduate College of Bowling Green State University in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY August 2015 Committee: H. Peter Lu, Advisor John Farver Graduate Faculty Representative Neocles Leontis John Cable © 2015 Qing Guo All Rights Reserved iii ABSTRACT H. Peter Lu, Advisor This dissertation presents our research work aiming at conformational manipulation of single enzyme protein molecules, performed by single molecule magnetic tweezers correlated with optical fluorescence spectroscopy. To experimentally investigate the enzyme-substrate interactions and the related conformational fluctuations, we have developed a new approach to manipulate the enzymatic conformation and enzyme-substrate interaction at the single-molecule level by using a combined magnetic tweezers and simultaneous fluorescence resonance energy transfer (FRET) spectroscopic microscopy. By a repetitive pulling-releasing manipulation of a Cy3-Cy5 dye labeled 6-hydroxymethyl-7,8-dihydropterin pyrophosphokinase (HPPK) molecule under the conditions with and without enzymatic substrates, we have probed and analyzed the enzymatic conformational dynamics. Our results indicate that the enzymatic conformational flexibility can be regulated by enzyme-substrate interactions: (1) the enzyme at its conformationperturbed state has less flexibility when binding substrates, and (2) substrate binding to the enzyme significantly changes the enzyme conformational flexibility, experimental evidence of so called entropy trapping in and enzyme-substrate reactive transition state. Furthermore, our results provide significant experimental analysis of folding-binding interactions of the enzymesubstrate interactions, and reveal the dynamic nature of the enzyme-substrate interactions. We also find supportive results from Steered Molecular Dynamics (SMD) Simulation, showing that in our studies, conformational manipulation by magnetic tweezers is able to distort the active domain of the enzyme molecules to an extent that significantly beyond thermal conformational fluctuations. iv Furthermore, we have also revealed the impact of partially unfolding the enzyme molecules on their activity by using single-molecule TIRF-magnetic tweezers spectroscopy to manipulate conformation of the enzyme molecules to a partially unfolded, yet not fully denatured condition. By conformationally distorting horseradish peroxidase (HRP) molecules via magnetic tweezers at the single molecule level, we successfully manipulated and examined the activity changes of the HRP catalyzed H2O2-Amplex Red reaction. We have observed significant tolerance of the enzyme activity to the enzyme conformation in its deformed or partially-unfolded states. We have identified that (1) enzymatic activity can be manipulated by our TIRF-magnetic tweezers at single molecule level; and (2) enzyme molecules in partially unfolded conformation are still capable of showing significant activity, although at a lower but measurable level, due to the enzymatic active site conformational fluctuation and substrate binding induced folding-binding conformational changes. We further provide our understanding of the enzyme behavior based on enzymatic conformational fluctuation, enzyme-substrate interactions, enzyme-substrate active complex formation, and protein folding-binding interactions. v To The Memory of My Dear Grandfather vi ACKNOWLEDGMENTS In my journey of becoming a Doctor of Philosophy at Bowling Green State University, I have received help and guide from many wise and friendly people. I am deeply grateful to them, and I appreciate their help and friendship. During the past six years of graduate study, if I have made any progress, either as a scientific researcher, or as a people in the society, I would like to attribute all of them to Dr. H. Peter Lu. I will be forever feeling in debt to him, for the countless support, patience, guidance, encouragement that Dr. Lu gave me. To me, Dr. Lu is the paragon in both being a successful scientist and being a socially welcomed people. He not only guides me in my work, but also offers me a lot of help in my life. It is him who improved my understanding of science. I would by no means to become Doctor without his chronic guidance and help. I am thankful to all my other committee members: Dr. Neocles Leontis, Dr. John Cable and Dr. John Farver for their precious time. I also want to acknowledge all the group members, both current members and past members from Dr. H. Peter Lu’s group, for setting a communication-free and hard-working environment. Especially, I want to thank Dr. Yufan He for his teaching and guidance to me these years. I also want to thank Jin Cao, Desheng Zheng, Yuanmin Wang and Zijian Wang for their friendship. I also want to say thank you to many faculty and staff at the Center for Photochemical Sciences and the Department of Chemistry: Nora Cassidy, Alita Frater, Charles Codding, and Doug Martin, Hilda Miranda, for their help. I would also like to thank my family for their love and support in these years. Without their understanding and support, I would never be able to enjoy working on science for so many years. vi TABLE OF CONTENTS Page CHAPTER I. INTRODUCTION .......................................................................................... 1 1.1 Introduction of Single Molecule Spectroscopy.............................................. 1 1.2 Introduction of Single Molecule Protein Conformational Dynamics ............ 4 1.3 Introduction of Single Molecule Studies of Enzyme ..................................... 10 1.4 Introduction of Magnetic Tweezers ............................................................... 12 1.5 Research Objective and Specific Aims, and Dissertation Overview ............. 17 1.6 References ...................................................................................................... 18 CHAPTER II. EXPERIMENT ............................................................................................. 25 2.1 2.2 Principles of Experimental Techniques ......................................................... 25 2.1.1 Principles of Confocal Microscopy ................................................... 25 2.1.2 Principles of Forster Energy Transfer (FRET) ................................. 28 2.1.3 Principles of Total Internal Reflection Microscopy (TIRFM) .......... 33 2.1.4 Signal Detection techniques: Introduction to APD and EMCCD...... 40 2.1.5 Basics of Magnetic Tweezers: Force Calibration .............................. 45 Experiment Details......................................................................................... 48 2.2.1 Experimental Setup of Single Molecule FRET Correlated with Magnetic Tweezers ........................................................................................................ 48 2.2.2 Experimental Setup of Single Molecule TIRFM Correlated with Magnetic 2.3 Tweezers ........................................................................................................ 50 2.2.3 Steered Molecular Dynamics (SMD) Simulation .............................. 51 Materials and Sample Preparation ................................................................. 52 vii 2.4 Selection of Magnetic Beads ......................................................................... 54 2.5 References ...................................................................................................... 56 CHAPTER III. MANIPULATING AND PROBING ENZYMATIC CONFORMATIONAL FLUCTUATIONS AND ENZYME-SUBSTRATE INTERACTIONS BY SINGLEMOLECULE FRET-MAGNETIC TWEEZERS MICROSCOPY .............................................................. 58 3.1 Introduction ................................................................................................... 58 3.2 Materials and Methods ................................................................................... 60 3.2.1 HPPK Protein ..................................................................................... 60 3.2.2 Sample Preparation ............................................................................ 62 3.2.3 Experimental System ......................................................................... 65 3.2.4 Force Calibration ............................................................................... 66 Results and Discussion .................................................................................. 67 3.3.1 FRET Measurement ........................................................................... 67 3.3.2 Repetitive Conformational Manipulation of Single HPPK Molecule 3.3 Observed by FRET Spectroscopy .................................................................. 3.3.3 Probing Conformational Flexibility of Single HPPK Protein Molecule by Single Molecule FRET-Magnetic Tweezers Spectroscopy ........................... 3.3.4 69 71 Conformational Dynamics Manipulation by Single Molecule FRET- Magnetic Tweezers Spectroscopy.................................................................. 76 3.4 Conclusion ..................................................................................................... 78 3.5 References ...................................................................................................... 79 CHAPTER IV. INTERROGATING THE ACTIVITIES OF CONFORMATIONAL DEFORMED ENZYME B BY SINGLE MOLECULE TIRF-MAGNETIC TWEEZERS viii MICROSCOPY ............................................................................................................ 89 4.1 Introduction .................................................................................................... 89 4.2 Materials and Methods ................................................................................... 89 4.2.1 Materials ............................................................................................ 92 4.2.2 TIRF Measurement ............................................................................ 93 4.2.3 Sample Preparation ............................................................................ 94 Results ............................................................................................................ 96 4.3.1 Single-Molecule TIRF Imaging Measurement of HRP Activity ....... 96 4.3.2 Analysis of Single-Molecule Activity Trajectories Measured under Force 4.3 Pulling and Releasing Conditions .................................................................. 4.3.3 97 Repetitive Force Pulling-Releasing Manipulation of Enzyme Conformation for Impacting Enzymatic Activity .......................................... 102 4.4 Discussion ...................................................................................................... 104 4.5 Conclusion ..................................................................................................... 107 4.6 References ...................................................................................................... 108 CHAPTER V. STEERED MOLECULAR DYNAMICS SIMULATION STUDIES OF THE CONFORMATIONALLY DEFORMED ENZYMES MANIPULATED BY SINGLE MOLECULE MAGNETIC TWEEZERS .............................................................................. 120 5.1 Introduction .................................................................................................... 120 5.2 Estimating Conformational Stretching Extent from HPPK Simulation ........ 121 5.3 SMD Simulation Study of HRP Protein Molecule ........................................ 125 5.3.1 SMD Simulation of HRP Protein Molecule in One Tethering Condition........................................................................................................ 125 ix 5.3.2 SMD Study on All Possible Stretching Type of HRP Protein Molecule ........................................................................................................ 129 5.3.3 5.4 Distortion in Unfolding Simulation ................................................... 131 References ...................................................................................................... 132 CHAPTER VI. DESIGN AND IMPLEMENTATION OF A QUADRUPOLE MAGNETIC TWEEZERS..................................................................................................... 133 6.1 History of Instrumental Design for Magnetic Tweezers................................ 133 6.2 The Multi-Channel Magnetic Tweezers ........................................................ 136 6.2.1 An Introduction of the Multi-Dimensional Magnetic Tweezers Setup in Our Lab .......................................................................................................... 136 6.2.2 An Improvement: Developing the New Generation Magnetic Tweezers ........................................................................................................ 140 6.3 References ...................................................................................................... 142 x LIST OF FIGURES Figure Page 1.1 Free Energy Diagram of Enzymatic Reaction ........................................................... 8 1.2 A Conceptual Scheme of the Sample System ............................................................ 14 2.1 Conceptual Figure of Confocal Microscopy .............................................................. 27 2.2 Principle of Förster Resonance Energy Transfer ....................................................... 30 2.3 The Distance-FRET Efficiency Relationship ............................................................ 32 2.4 Spectrum Profile of Chromophore Molecule Cy3 and Cy5....................................... 33 2.5 A Conceptual Figure of Total Internal Reflection Phenomenon ............................... 35 2.6 A Conceptual Scheme of Evanescent Wave .............................................................. 37 2.7 Conceptual scheme of total internal reflection microscopy (TIRFM) ....................... 39 2.8 Principle of Photodiode Module used in our experiment .......................................... 42 2.9 Principle of Electron Multiplied Charge Coupled Device (EMCCD) ....................... 44 2.10 Magnetic field-Distance curve of the magnet used in experiment ............................ 47 2.11 Experimental setup of single molecule FRET-Magnetic Tweezers spectroscopy..... 49 2.12 Experimental setup of single molecule TIRF-Magnetic Tweezers spectroscopy ...... 50 2.13 A conceptual scheme of single molecule protein immobilization method ................ 53 2.14 Force-velocity response of magnetic beads in different size ..................................... 55 3.1 Structural information of HPPK molecule. ................................................................ 61 3.2 Preparation of single molecule HPPK sample. .......................................................... 63 3.3 A conceptual scheme of the experimental system ..................................................... 65 3.4 Single-molecule FRET data of a Cy3-Cy5 labeled HPPK in experiment ................. 68 3.5 Repetitive force pulling and releasing manipulation of Single HPPK molecules ..... 70 xi 3.6 Perturbing and characterizing enzyme-substrate binding interaction by single-molecule FRET magnetic tweezers microscopy ....................................................................... 3.6 73 Conformational fluctuation rate distributions calculated from autocorrelation analysis of HPPK with substrate ATP and HP added. ................................................................. 75 4.1 A conceptual scheme of our experimental system..................................................... 92 4.2 Preparation of single molecule HRP sample ...................................................... 95 4.3 Single-turnover detection of HRP enzyme catalysis ................................................. 97 4.4 Histogram results of turnover events from 30 individual HRP molecules ................ 98 4.5 Analysis of the relationship between turnover event, mean waiting time and product burst of single HRP molecules ............................................................................................ 101 4.6 Response of HRP enzymatic activity to repetitive magnetic pulling force ............... 103 4.7 Conceptual scheme of conformational fluctuation of single enzyme protein when being deformed by external force ........................................................................................ 107 5.1 SMD simulation of HPPK molecule pulling by magnetic tweezers. ......................... 124 5.2 SMD simulation results show the scheme of the distortion of active site when the protein is pulling by magnetic tweezers ................................................................................. 127 5.3 Three residue pairs to illustrate distortion on the active domain when an HRP molecule is stretched by magnetic tweezers ................................................................................. 130 5.4 Active site conformational distortion in larger unfolding situation ........................... 131 6.1 A conceptual scheme of the quadrupole magnetic tweezers setup ............................ 137 6.2 Conceptual scheme of an experiment testing the function electromagnet poles ....... 138 6.3 A testing experiment examination the electromagnet functions. ............................. 139 6.4 A circuit diagram of the new controlling system for the quadrupole electromagnet. 141 1 CHAPTER I. INTRODUCTION This chapter is dedicated to the introduction of single molecule studies of protein conformational dynamics and a brief introduction of single molecule magnetic tweezers (MTW). 1.1 Introduction of Single Molecule Spectroscopy The capability of probing and manipulating molecules at single molecule level and even to manipulate chemistry reactions by probing chemical bonds has been a chronic dream by chemists. With the development of lasers and microscope techniques, it started to become possible in experiment in late 20th century. Single molecule spectroscopy originates from the studies on solid state science, which used fluorescence spectroscopy to detect a single molecule in crystalline or amorphous solids as a probe of local structure and dynamics in solids. The first successful single molecule experiment has been achieved by Moerner’s group back to 1989, using sensitive doubly modulated absorption approach to detect a single molecule in solids at low temperature.1 By 1990, a single molecule observation in liquid has been achieved;2 in 1993, Betzig and Chichester have firstly observed immobilized single molecule at room temperature by near-field scanning microscope.3 Afterward, single molecule spectroscopy has already been regarded as a potentially rich field and powerful approach for biophysics research, although the experimentally technical difficulties of near field scanning method still limit the application of single molecule methods. From 1994 to 1997, imaging single molecule at room temperature using far field microscopy and Raman spectroscopy have been achieved.4,5 In 1998, Xie and Lu firstly applied single molecule room temperature 2 fluorescence spectroscopy in enzymatic dynamics study, which finally started a new era using single molecule approaches in biophysics research.6 Afterward, single molecule techniques have been widely used in extensive studies on biophysics field. Depending on research focus, those works since then can be sorted into two large directions. One of them is focused on imaging and spectra analysis, such as Surface Enhanced Raman Spectroscopy (SERS),4 Förster Resonance Energy Transfer (FRET) spectroscopy,7 and more recent studies on super-resolution imaging methods for biological systems such as Stimulated Emission Depletion (STED) Microscopy, Photoactivated Localization Microscopy (PALM) and Stochastic Optical Reconstruction Microscopy (STORM).8‐10 In the other direction, scientists are more interested in mechanical characters of biomolecules, and hence developing a lot of techniques to apply force on target sample at single molecule level, such as Atomic Force Microscopy (AFM),11‐12 Optical Tweezers developed by Chu, Bustamante and Block,13‐15 Magnetic Tweezers developed by Bustamante and Bensimon16,17, Anti-Brownian Electrokinetic Trap developed by Moerner18 Biomembrane Force Probe developed by Evans,19 etc. A lot of theoretical studies also spring out to enhance the single molecule field, such as protein folding-unfolding theory developed by Wolynes and Onuchic,20, 21 and single molecule level enzymatic reaction theory developed by Xie and Lu, etc. 4,22 Single molecule approaches can provide new information that cannot be obtained from traditional ensemble level experiments in chemical and biological research. In ensemble level measurements, which means that individual behavior of molecules are indistinguishable, experimentally observed parameters are usually average characteristics. By removing ensemble averaging effects, single molecule techniques are scientifically 3 valuable mainly in three aspects. Firstly, single molecule measurements generate frequency histogram of the distribution of experimentally observed values. Statistical indicators of these distributions will bring more information, i.e. multi-peak histogram which reflects multiple intermediate states; the shape of the histogram which reflects fluctuation flexibility of respective parameters. Such characters are especially important when the sample systems are heterogeneous. Secondly, single molecule methods make post-synchronization of time-dependent processes with many molecules involved unnecessary. For a process such as enzymatic reaction or protein-protein interaction which involves multiple different states, when we measure them at ensemble level, at any given time, the specific states of molecules in our detection signal will be averaged out and cause loss of information from observation of molecule intermediate states. However, if we can monitor one molecule at a time with proper time resolution, at any given time, its specific states will be recorded without any averaging out effect, and hence give us more information of the sample states on time domain. As a result, more dynamical information can be obtained in single molecule measurements. For example, in single molecule detections, since all the signals are from one same molecule, we can apply correlation analysis to analyze the fluctuation rate of certain states of the molecule, or we can even analyze time-involved disorder of parameters of one single molecule.23 Thirdly, in single molecule observation, new effects can be discovered, or even become diagnostics of molecule system. For example, absorption spectra frequency of single molecule has been found to be an indicator of molecular local spatial environment;24, 25 Fluctuation rate from autocorrelation analysis can be a diagnostic approach to reveal process which involves multiple intermediate states. 4 Those advantages make single molecule spectroscopy become especially powerful for biophysics studies. On one hand, biological process such as protein foldingunfolding or enzyme catalysis process at molecule level are more dynamic rather than static, which means time-dependent fluctuation plays a key role in those processes. On the other hand, single molecule techniques also provide us detailed structure detecting methods and even manipulation methods of biological systems such as DNA or protein molecules, membrane systems. Moreover, many biophysical processes such as DNA translation, gene expression, or transportation occurring on membranes involve very few molecules or even single molecule.26 In later chapters, our discussion will mainly focused on protein conformational dynamics and structure-function relationship of enzyme molecules at single molecule level. 1.2 Introduction of Single Molecule Protein Conformational Dynamics Protein are participants in various types of processes in living organisms such as metabolic reactions, gene expression, response to external stimulation, etc. and hence constitute an active research area in life science. As a result, understanding the structure and dynamics of protein molecules and the mechanism of their functions has been intriguing scientists’ interest for a long time. The prototype of modern idea of protein structure has been developed since 1950s.27 The first protein having its three-dimensional structure resolved is myoglobin, achieved by Kendrew in 1958 using X-Ray Crystallography. In 1965, Blake resolved the structure of lysozyme, making it the first enzyme and the second protein molecule having its structure revealed. Since then, by the help of several techniques such as X-Ray Crystallography, Nuclear Magnetic Resonance (NMR) and Cryo-Electron Microscopy (Cryo-EM), more than 35,000 distinct protein 5 sequences have been resolved. Nowadays, crystal structures of many complex protein molecules such as NMDA receptors can be decoded in experiments.28 In biophysics and biochemistry studies today, a major concerned topic is the structure-function relationship. The structure of protein molecules are heterogeneous biopolymer which is formed by folding a particular linear sequence consisting of 20 naturally occurring amino acids. Once the sequence is determined, the amino-acids chain will be regulated by many different types of non-covalent force, including hydrogen bonding, hydrophobic forces, electrostatic forces, van der Waals forces, etc. and therefore folded into a 3-D structure spontaneously. Such 3-D structures, although recently found to be fluctuating in natural condition, are called stable conformation of the protein molecules. For protein molecules, it has been found out that structure and function of protein molecules are intimately related. For example, specific conformational recognition processes are involved in antibody-antigen binding interaction; enzymesubstrate conformational docking is the key step in enzymatic catalytic function; neurotransmitters rely on conformation-specific binding of receptors that ties on membrane protein of the target cell to achieve neuro-signal transportation, etc. Protein conformation is also dynamic rather than static. In real biological environment, conformations of protein molecules are not limited in one certain configuration, but fluctuate all the time. For example, internal motions of enzyme molecules have been found fluctuating at different time scales ranging from pico-seconds to seconds. Furthermore, in biological processes such as protein-ligand interactions or enzyme-substrate interactions, protein molecules undergo a lot of intermediate states with different conformations, while time dependent study on conformation facilitate us to 6 understand mechanisms of those processes. Such dynamic characteristics of protein conformation allow us to reveal rich information from time-resolved single molecule spectroscopy. For example, in protein folding-unfolding studies, each conformation of a given protein molecule reflects different potential energy; the landscape consisting of those different energies as a function of different conformation reveals the folding probability of protein molecules.20 In single molecule enzymology studies, the conformational fluctuation rate of enzyme molecules which can be obtained from autocorrelation analysis of experimental time trajectories can even reveal the existence of multiple intermediate states which enzyme molecules undergo during enzymatic reactions.29 Based on previous knowledge of crystal structures of protein molecules, extensive single-molecule studies of time-dependent information of protein conformation have come out. In recent decades, both theoretical and experimental research works have been done to study dynamic character of protein conformation in many different processes. For example, theoretical models based on the concept of energy landscape describing protein folding-unfolding process have been developed by Wolynes and Onuchic,20,21 experimentally observations of protein folding-unfolding and folding-binding process have been studied by many groups,30,31 intermediate states of enzyme protein during its catalytic process have been revealed by Lu since 2004, etc.32 Recently there is even a research field named independent disordered protein which is specifically focused on conformational fluctuation regulation of protein molecules. One type of proteins that are especially important is enzymes. Enzymes are protein molecules that can speed up chemical reactions by thousands or even millions of 7 times by changing the energy barrier, or so-called activation energy to accelerate the formation of intermediate states of given reactions. Typically, such processes are accomplished via specific binding process between substrate molecules and active site on enzyme molecules. The term ‘active site’ refers to a part of an enzyme molecule, taking charge of binding with substrate to catalyze chemical reactions. The active site on an enzyme molecule typically consists of a few amino acid residues which directly participate in the recognition of substrate molecules to initiate the catalytic reaction mechanism. The catalysis process in a chemical reaction is usually triggered by a collision between a substrate and the active site, which then evolved into a specific binding process between the substrate and the active site. Some enzymes have their active sites accessible in their 3-D conformations; some enzymes have their active sites buried inside their 3-D structure, requiring conformational change to allow the substrate to access the active site. The ability of enzyme to increase chemical reaction or biological reaction rate and the specificity of enzyme function intrigues extensive interests of chemists for a long time. From ensemble level experiments, kinetic model describing enzymatic reaction has been established a hundred years ago.33,34 Models describing the kinetics of an enzymatic reaction can be shown in equation 1.1. k k3 kcat 1 ZZZ X E + S YZZ → EP ⎯⎯→ E+P Z ES ⎯⎯ k 2 (1.1) In equation 1.1, E stands for enzyme, while S and P are short for substrate and products respectively. Rate constant of each steps are noted as k1 to k3 and kcat. An enzymatic reaction is a process that including at least three steps: the first step is the enzyme 8 substrate compleex formation n, while the second s step iis chemical rreaction by bbreaking andd form ming chemicaal bonds, and d the final step will be prroduct releassing. The ennergy change du uring an enzy ymatic reacttion is descriibed in figurre 1.1. Figurre 1.1. Free energy diagrram showing g that enzym me increase cchemical reaaction by lowerring activatio on free energ gy barrier. The T blue currve indicatess an enzymattic reaction, whilee the black curve indicates the same chemical reaaction withoout enzyme ccatalysis. Acron nyms stand for: E for en nzyme; S forr substrate; P for productt; ES for enzzymesubsttrate complex x; EP for enzyme-produ uct complex. ES* stands ffor the transsition state. ΔG*caat and ΔG*uncat are activaation free eneergy of the eenzymatic reeaction with and without enzym me respectiv vely. 9 Theory to describe the overall rate of enzymatic reaction has also been established by Michaelis and Menton since 1913, which can be summarized in equation 1.2.34 v = kcat [ E ]0 [S ] K M + [S ] (1.2) In equation 1.2, v stands for the reaction rate, [E]0 is the enzyme concentration, kcat is the maximum number of substrate molecules that being converted into product during the reaction per enzyme molecule per second. KM indicates the substrate concentration when the reaction rate reaches its half-maximum value. [E] and [S] stands for concentration of enzyme and substrate respectively. Despite kinetic model has been well-developed to describe mechanism of enzymatic reactions at ensemble level, structure-function question for enzyme molecules remains as a challenge. Since late 1990s, with the development of single molecule techniques, research on enzymatic reaction has also entered a new level. Fluorescencebased optical observation methods have been developed, which allow us to monitor timedependent structure information of single enzyme protein molecule or reaction turnover events; theoretical models for enzyme at single molecule level has been developed in recent years too.22 Although some conventional approaches such as NMR and XRD can resolve enzyme protein conformation in crystal structure form, it has to rely on recently developed single molecule methods to probe the impact of enzyme conformation to its activity during reactions, which will be discussed in the following section. 10 1.3 Introduction of Single Molecule Studies of Enzyme Using single molecule approaches, such as single molecule fluorescence spectroscopy and single molecule force spectroscopy, we are able to interrogate conformational dynamics and the associated functions of enzyme molecules at single molecule level. The advantages of single molecule techniques which have been discussed in section 1.1 make recent research of enzymatic reactions rely on single molecule approaches to answer the structure-function questions of enzyme molecules and to reveal their dynamic characteristics: For a given enzymatic reaction, what is happening to enzyme molecules when they perform catalytic function? How do substrate ligands affect or even regulate enzyme-substrate binding events? What does the conformation of enzyme molecules at intermediate states look like? Is there one single intermediate or multiple intermediate states of enzyme-substrate complex during a reaction? What is the impact from conformational fluctuation of enzyme molecules on their functions, and can we manipulate those conformational fluctuations? Compared to traditional ensemble level experiments studying enzymatic reactions, single molecule level experiments allow us to directly probe the formation of enzyme–substrate reactive complex or even the conformation of active sites on enzyme protein molecules, and to answer those questions. In this dissertation, our research works are focused on studying conformational dynamics of enzyme protein molecules at single molecule level. Enzyme-substrate interactions determine the formation of enzyme-substrate complex which is the initial triggering step of an enzymatic reaction. Understanding the enzyme-substrate interaction will be helpful in characterizing formation of enzymatic transition states that defines the reaction pathway, energetics, and the dynamics. A critical factor determining the rate of 11 enzyme–substrate interactions is enzymatic conformational change. Protein conformation has been found to be a critical impact factor in determining binding affinity toward ligands.35 As a result, conformational regulation can significantly affect active site-substrate binding process. For example, conformational stability of tertiary structure has been found to have important influence on enzymatic activity;36,37 by manipulating conformation of enzyme molecules, even controlling enzyme-substrate interaction is possible.38 To understand the role of conformational change of enzyme molecules in catalytic process, many different models have been developed. Back in 1894, Fisher proposed a lock-key hypothesis, qualitatively describing the high selectivity of enzyme-substrate interactions using a metaphor which is ‘the specificity of the combination of a key and respective lock’.39 In 1958, Koshland firstly suggested an ‘induced-fit’ theory, claiming that the binding interaction between an enzyme protein and substrate molecule induces conformational change to the enzyme protein. By mid 1960s, a ‘conformational selection’ paradigm was proposed, stating that conformations of enzyme molecules are fluctuating all the time, and substrate ligands will select those conformations that are compatible with enzyme-substrate binding, resulting in a conformational distribution shift to those reaction-favored ones.40,41 In our studies in this dissertation, we also try to interrogate the impact of enzyme conformation to their catalytic function. We developed magnetic tweezers correlated with single molecule fluorescence spectroscopy for the purpose of probing the enzyme protein conformational dynamics and the associated enzymatic function. Firstly, we successfully achieved repetitive conformational manipulation of single enzyme protein 12 molecule. Later, using this conformational manipulation method, we try to interrogate the conformational selection mechanism by probing the conformation flexibility under different conditions aiming at revealing the influence of enzyme-substrate interactions on conformational dynamics of enzyme molecules, and to discover dynamic nature of the enzymatic active transition state formation process. We further make the attempt to interrogate structure-function relationship at molecule level in enzymatic reactions. Traditional enzymatic stability studies focused on ensemble level activity of enzyme at different physical conditions or chemical environment without probing corresponding change in conformation of enzyme molecules. Hence, the impact to enzymatic activity from partial conformational change, which is the condition that enzyme molecules are not unfold or denatured, but only with some stretching or distortion on its conformation remains unclear. Using single molecule magnetic tweezers as conformational probe to manipulate enzyme activity at single molecule level with simultaneous optical observation of turnover events, we are trying to answer this question. 1.4 Introduction of Magnetic Tweezers Force is a key parameter which is involved in all types of biological processes. For example, the motions of motor proteins generate biological force;42 Ligand-receptor recognition processes involve binding force;43 Protein folding-unfolding events can be easily affected by external force.44 There are also some force-driven processes, such as gene expression or cellular motions.45 There are even many proteins having mechanical functions , such as cytoskeletal proteins or muscle proteins.46 Hence, the capability to 13 measure and to apply force onto single molecules in order to probe those fundamental processes becomes a major concern in biophysical and biochemistry studies. Applying molecular level force to manipulate molecules that involved in biological processes typically require mechanical force at 10-12 Newton (pico-newton, or pN) scale. Meanwhile, such manipulation approaches also need to achieve a high spatial precision at 10-9 meter (nm) scale. In the last 30 years, there are plenty of techniques developed for this purpose, the most successful and thus commonly used techniques are Atomic Force Microscopy (AFM), Optical Tweezers, Magnetic Tweezers, Biomembrane Force probe (BFP) and Microneedles.11‐19,47 In our studies, we use magnetic tweezers as the manipulation approach to apply force on single protein molecules. Magnetic force has been used in biological and medical field for a long time. For example, magnetic resonance imaging (MRI) has been widely used as a clinical diagnostic method; magnetic force for drug targeting has also been developed since 2006.48 The concept of magnetic tweezers is to employ magnetic field to apply force through magnetic nanoparticles as force sensor conductor onto target molecules. A classical way of doing single molecule magnetic tweezers experiment is shown in figure 1.2. A target bio-molecule sample, which can be either single DNA or protein molecule or even biological specimen, is immobilized on the glass cover slip tethered by covalent bond. And in the other end of the sample molecule, a paramagnetic nanoparticle is linked via specific covalent bond. This paramagnetic particle will serve as the force sensor, conducting force applied by external magnetic field to apply force onto the sample molecule. The applied force can be regulated by controlling the magnetic field. To apply magnetic field, either permanent magnet or electromagnet can be used, while the field 114 can be b tuned by either e spatiallly controllin ng the positioon of magneet or controllling the current applied on n electromag gnet. Figurre 1.2. A conceptual sch heme of the sample s systeem in magneetic tweezerss experimentts. In briief, we tether a single prrotein molecu ule to a moddified glass ccoverslip at oone end by TESP PA-DMS lin nkers and bou und to a super-paramagnnetic bead att the other ennd via biotinn- 15 streptavidin specific binding. When external magnetic field is applied, the protein molecule will be pulled via sensing the force through the magnetic bead. The earliest single molecule work applying magnetic tweezers to manipulate biomolecules can be traced back to 1992, which is the study by Bustamante using magnetic beads to measure the elasticity of single DNA molecules. The term ‘magnetic tweezers’ has not been widely used until 1996, when Bensimon used magnetic force to wring out DNA molecules. Since then, magnetic tweezers have been widely applied in many different biophysical research works as a molecular force manipulation approach. There are extensive works improving the setup of magnetic tweezers;49 many different DNA molecules have been characterized in their mechanical parameters using magnetic tweezers;50 Some researchers try to use magnetic tweezers as a force calibration approach or a force clamp technique;51 Some studies trying to use magnetic force for cellular nanoparticle transportation have also been achieved.52 Why do we need to develop magnetic tweezers? The answer is that as a molecule level force probe, magnetic tweezers have some significant advantages compared to other alternative methods such as AFM and optical tweezers. Firstly, it can apply pulling force either small as sub-picoNewton49 or large as close to nanoNewton. In contrast, AFM can only generate pulling force not smaller than 5 pN until 2012, with a complicated modified experiment setup and micro-machine modified cantilever. The reason lies in different working mechanism of these approaches. In experiments, AFM needs real-time response of the physical position of AFM cantilever to calibrate real-time applied force. Hence, the spring constant of the AFM cantilever will become the limiting factor of how small the force can be achieved. On the other hand, magnetic tweezers do not require any 16 corresponding physical feedback observation to provide force information in experimental measurements, although force calibrations need to done independently. Details of force calibration in magnetic tweezers experiment will be discussed in Chapter II. The second advantage is that direct physical contact or chemical contact to target molecules is not necessary in magnetic tweezers experiments. Magnetic tweezers indeed require a magnetic nanoparticle, or named magnetic beads to direct tethered onto sample molecules as a force sensor to conduct applied force by magnetic field. However, in experimental measurements, it still allow a remote control, or ‘controlling via field’ type force to be applied. As a result, in magnetic tweezers experiments, any inaccuracy from direct physical contact of force applying part in instrument such as AFM cantilever vibration or displacement can be avoided. Also remote control significantly facilitates the experiment design and reduces complexity for any correlated measurement. The third character of magnetic tweezers as a single molecule level force manipulation method is that any photon-damage to the sample or a photon induced background noise to a correlated simultaneous single-molecule spectroscopic measurement can be avoided. Compared with optical tweezers, which always require lasers at infrared region and high power to create optical potential field trap to manipulate protein spatial position, magnetic tweezers is beneficial in this aspect not only by avoid of inducing photon damage or photon noise, but also has the advantage by not to induce any heat as side effect of high power lasers into experimental systems. 17 Finally, magnetic tweezers allows manipulating large number of molecules simultaneously as long as those molecules have paramagnetic micro beads tethered on them. In recent years, using an improved modified technique, AFM can also apply force on multiple protein molecules at the same time.53 However, considering the instrumental complexity and force scale limit of AFM, magnetic tweezers is still a unique technique to apply force on multiple molecules. These specificities make the magnetic tweezers approach promising for protein conformational dynamics studies, especially for our research purpose in this dissertation. 1.5. Research Objective and Specific Aims, and Dissertation Overview Previously, research works using magnetic tweezers to study single bio-molecules majorly focused on manipulating DNA topological structure, or resolving unfolding dynamics of poly-protein molecules. Manipulating single protein conformation and to further probe the impact of structure change of protein to its function using magnetic tweezers remains unchallenged. The objective of our research is using magnetic tweezers correlated with single molecule fluorescence optical spectroscopy to manipulate conformation of single protein molecule, and to probe structure-function relationship of enzyme protein at single molecule level. This dissertation consists of six chapters. Chapter I discussed about scientific motivation of the research work in this thesis. Chapter II described instrumental setups in our experiments and some necessary background knowledge including principles of some experimental techniques used in our studies. Chapter III presents our work using a combined magnetic tweezers and simultaneous fluorescence resonance energy transfer 18 (FRET) spectroscopic microscopy to manipulate the conformation of a Cy3-Cy5 dye labeled 6-hydroxymethyl-7,8-dihydropterin pyrophosphokinase (HPPK) molecule and enzyme-substrate interaction at single-molecule level. Chapter IV shows that by applying conformational distortion of horseradish peroxidase (HRP) molecules via magnetic tweezers at single molecule level, we successfully manipulated and examined the activity changes of HRP catalyzed H2O2-Amplex Red reaction. We made further theoretical studies using Steered Molecular Dynamics (SMD) simulation method to have a detailed understanding about the impact on enzyme active domain when external force is applied via magnetic tweezers in chapter V. In chapter VI, we discussed a newly developed technical improvement for higher controlling ability for next generation of magnetic tweezers. 1.6. References 1 Moerner, W.E.; Kador, L. Finding a Single Molecule in a Haystack: Optical Detection and Spectroscopy of Single. Absorbers in Solids. Anal. Chem., 1989, 61 (21), pp 1217A–1223A. 2 Shera, E.B.; Seitzinger, N.K.; Davis, L.M.; Keller, R.A.; Soper, S.A. Detection of Single Fluorescent Molecules. Chem. Phys. Lett. 1990, 174 (6), 553-557. 3 Betzig, E.; Chichester, R.J. Single Molecules Observed by Near-Field Scanning Optical Microscopy. Science 1993, 262, 1422-1425. 4 Nie, S.; Chiu, D. T.; Zare, R. N. Probing Individual Molecules with Confocal Fluorescence Microscopy. Science 1994, 266, 1018-1021. 5 Macklin, J. J.; Trautman, J. K.; Harris, T. D.; Brus, L. E. Imaging and Time-Resolved Spectroscopy of Single Molecules at an Interface. Science 1996, 272, 255-258. 19 6 Lu, H.P.; Xie, X.S. Single-Molecule Enzymatic Dynamics. Science, 1998, 282(5395):1877-82. 7 Ha, T.; Enderele, D.F.; Ogletree, D.S.; Chemla, D.S.; Selvein, P.R.; Weiss, S. Probing the interaction between two single molecules: Fluorescence resonance energy transfer between a single donor and a single acceptor. Proc. Natl. Acad. Sci. USA 1996, 93, 6264-6268. 8 Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging Intracellular Fluorescent Proteins at Nanometer Resolution. Science, 2006, 313 ( 5793) 1642-1645. 9 Hell, S.W.; Wichmann, J. Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 1994, 19(11) 780-782. 10 Rust, M.J.; Bates, M.; Zhuang, X. Stochastic optical reconstruction microscopy (STORM) provides sub-diffraction-limit image resolution. Nat. Methods. 2006, 3(10): 793–795. 11 Rief, M.; Gautel, M.; Oesterhelt, F.; Fernandez, J.M.; Gaub, H.E. Reversible unfolding of individual titin immunoglobulin domains by AFM. Science. 1997 276(5315):1109-12. 12 Rief, M.; Oesterhelt, F.; Heymann, B.; Gaub, H.E. Single Molecule Force Spectroscopy on Polysaccharides by Atomic Force Microscopy. Science. 1997 275(5304): 1295-1297. 20 13 Smith, S.B.; Cui, Y.; Bustamante, C. Overstretching B-DNA: The Elastic Response of Individual Double-Stranded and Single-Stranded DNA Molecules. Science. 1994 271(5250): 795-799. 14 Ashkin, A.; Dziedzic, J.M.; Bjorkholm, J.E.; Chu, S. Observation of A Single-Beam Gradient Force Optical Trap for Dielectric Particles Opt. Lett. 1986, 11(5) 288-290. 15 Svoboda, K.; Schmidt, C.F.; Schnapp, B.J.; Block, S.M. Direct Observation of Kinesin Stepping by Optical Trapping Interferometry. Nature 1993, 365 (6448), 721727. 16 Smith, S.B.; Finzi, L.; Bustamante, C. Direct Mechanical Measurements of The Elasticity of Single DNA Molecules by Using Magnetic Beads. Science. 1992 258(5085): 1122-1126. 17 Strick, T.R.; Allemand, J.F.; Bensimon, D.; Bensimon, A.; Croquette, V. The Elasticity of A Single Supercoiled DNA Molecule. Science. 1996 271(5257): 18351837. 18 Cohen, A.E.; Moerner, W.E. The Anti-Brownian Electrophoretic Trap (ABEL trap): Fabrication and Software. Biomedical Optics 2005, 296-305. 19 Evans, E. Probing the Relation Between Force—Lifetime—and Chemistry in Single Molecular Bonds. Annu. Rev. Biophys. Biomol. Struct. 2001, 30, 105-128. 20 Bryngelson, J.D.; Jose Nelson Onuchic, J.N.; Socci, N.D.; Wolynes, P.G. Funnels, Pathways, and the Energy Landscape of Protein Folding: A Synthesis. Proteins: Struct., Funct., Bioinf. 1995, 21(3), 167-195. 21 Frauenfelder, H.; Sligar, S.G.; Wolynes, P.G. The Energy Landscapes and Motions of Proteins. Science. 1991 254 (5038): 1598-1603. 21 22 Kou, S.C.; Cherayil, B.J.; Min, W.; English, B.P.; Xie, X.S. Single-Molecule Michaelis−Menten Equations. J. Phys. Chem. B, 2005, 109 (41), 19068–19081. 23 Xie, X.S. Single-Molecule Approach to Dispersed Kinetics and Dynamic Disorder: Probing Conformational Fluctuation and Enzymatic Dynamics. J. Chem. Phys. 2002, 117(24), 11024-11032. 24 Kneipp, K.; Wang, Y.; Kneipp, H.; Perelman, L.T.; Itzkan, I.; Dasari, R.R.; Feld, M.S. Single Molecule Detection Using Surface-Enhanced Raman Scattering (SERS). Phys. Rev. Lett. 1997, 78, 1667. 25 Lu, H.P.; Xie, X.S. Single-Molecule Spectral Fluctuations at Room Temperature. Nature 1997, 385, 143 – 146. 26 Tamarat, Ph.; Maali, A.; Lounis, B.; Orrit, M. Ten Years of Single-Molecule Spectroscopy. J. Phys. Chem. A, 2000, 104(1), 1-16. 27 Pauling, L.; Corey, R.B.; Branson, H.R. The Structure of Proteins; Two HydrogenBonded Helical Configurations of the Polypeptide Chain. Proc. Natl. Acad. Sci. USA 1951, 37(4), 205-211. 28 Karakas, E.; Furukawa, H. Crystal Structure of a Heterotetrameric NMDA Receptor Ion Channel. Science, 2014, 344, 992-997. 29 Wang, Y.; Lu, H.P. Bunching Effect in Single-Molecule T4 Lysozyme NonEquilibrium Conformational Dynamics under Enzymatic Reactions. J. Phys. Chem. B, 2010, 114, 6669-6674. 30 Schuler, B.; Lipman, E.A.; Eaton, W.A. Probing the Free-Energy Surface for Protein Folding with Single-Molecule Fluorescence Spectroscopy. Nature 2002, 419 (6908), 743-747. 22 31 Deniz, A.A.; Laurence, T.A.; Beligere, G.S.; Dahan, M.; Martin, A.B.; Chemla, D.S.; Dawson, P.E.; Schultz, P.G.; Weiss, S. Single-Molecule Protein Folding: Diffusion Dluorescence Resonance Energy Transfer Studies of the Denaturation of Chymotrypsin Inhibitor 2. Proc. Natl. Acad. Sci. USA 2000, 97(10), 5179–5184. 32 Chen, Y.; Hu, D.; Vorpagel, E.R.; Lu, H.P. Probing Single-Molecule T4 Lysozyme Conformational Dynamics by Intramolecular Fluorescence Energy Transfer. J. Phys. Chem. B, 2003, 107 (31), 7947–7956. 33 Fersht, A. R. Structure and Mechanism in Protein Science: a Guide to Enzyme Catalysis and Protein Folding, Freeman Publishing, 1999. 34 Michaelis, L.; Menten, M.L.; Die kinetik der invertinwirkung. Biochem. z, 1913, 49, 333–369. 35 Gilson, M.K.; Zhou, H. Calculation of Protein-Ligand Binding Affinities. Annu. Rev. Biophys. Biomol. Struct. 2007, 36: 21-42. 36 Klink, T.A.; Raines, R.T. Conformational Stability Is a Determinant of Ribonuclease A Cytotoxicity. J. Biol. Chem. 2000, 275, 17463–17467. 37 Iyer, P.A.; Ananthanarayan, L. Enzyme Stability and Stabilization—Aqueous and Non-Aqueous Environment. Process Biochem. 2008, 43, 1019–1032. 38 Guo, Q.; He, Y.; Lu, H.P. Manipulating and probing enzymatic conformational fluctuations and enzyme-substrate interactions by single-molecule FRET-magnetic tweezers microscopy. Phys. Chem. Chem. Phys. 2014, 16: 13052-13058. 39 Fischer, E. Influence of Configuration on the Action of Enzyme. Reports of The German Chemical Society, 1894, 27(3): 2985-2993. 23 40 Changeux, J-P. Jean Thiéry, J.; Tung, Y.; Kittel, C. On the Cooperativity of Biological Membranes. Proc. Natl. Acad. Sci. USA 1967, 57(2), 335–341. 41 Straub, F. B.; Szabolcsi, G. O dinamicseszkij aszpektah sztukturü fermentov. (On the dynamic aspects of protein structure) In: Molecular Biology, Problems and Perspectives, (Ed.: Braunstein, A. E., Russian), Izdat. Nauka, Moscow, 1964, pp. 182–187. 42 Finer, J. T.; Simmons, R. M.; Spudich, J. A. Single Myosin Molecule Mechanics: PicoNewton Forces and Nanometer Steps. Nature, 1994, 368, 113-119. 43 Florin, E.L.; Moy, V.T.; Gaub, H.E. Adhesive Forces Between Individual LigandReceptor Pairs. Science 1994, 264, 415-417. 44 Fernandez, J.M.; Li, H.; Force-Clamp Spectroscopy Monitors the Folding Trajectory of a Single Protein. Science 2004, 303 (5664): 1674-1678. 45 Benoit, M.; Gabriel, D.; Gerisch, G. Gaub, H.E. Discrete Interactions in Cell Adhesion Measured by Single-Molecule Force Spectroscopy. Nature Cell Biol. 2000, 2, 313 – 317. 46 Tskhovrebova, L.; Trinick, J; Sleep, J.A.; Simmons, R.M. Elasticity and Unfolding of Single Molecules of the Giant Muscle Protein Titin. Nature 1997, 387, 308-312. 47 Kishino, A.; Yanagida, T. "Force measurements by micromanipulation of a single actin filament by glass needles". Nature 1988, 334 (6177): 74–76. 48 Haacke, E.M.; Brown, R. W.; Thompson, M.R.; Venkatesan, R. Magnetic Resonance Imaging: physical principles and sequence design, Wiley-Liss, New York, 1999. 49 Kollmannsberger,P.; Fabry, B. High-force magnetic tweezers with force feedback for biological applications. Rev. Sci Instrum. 2007, 78, 114301. 24 50 Lionnet, T.; Joubaud, S.; Lavery, R.; Bensimon, D.; Croquette, V. Wringing Out DNA. Phys. Rev. Lett. 2006, 96, 178102. 51 del Rio, A.; Perez-Jimenez, R.; Liu, R.; Roca-Cusachs, P.; Fernandez, J.M.; Sheetz, M.P. Stretching Single Talin Rod Molecules Activates Vinculin Binding. Science. 2009 323 (5914): 638-641. 52 Bausch, A.R.; Moller, W.; Sackmann, E. Measurement of Local Viscoelasticity and Forces in Living Cells by Magnetic Tweezers. Biophys. J. 1999, 76(1) 573–579. 53 Gumpp, H.; Puchner, E.M.; Zimmermann, J.L.; Gerland, U.; Gaub, H.E. Blank, K. Triggering Enzymatic Activity with Force. Nano. Lett. 2009, 9 (9):3290-3295. 25 CHAPTER II. EXPERIMENTS This chapter is dedicated to the description of experimental techniques in our single molecule magnetic tweezers (MTW) studies and the sample preparation procedures in our experiments. 2.1 Principles of Experimental Techniques 2.1.1 Principles of Confocal Microscopy The prototype of the concept of ‘confocal’ can be traced back in 1940, when Hans Goldmann developed a slip lamp system for eye examination.1 The so-called confocal microscope came out firstly at 1950s, developed by Marvin Minsky, who was a postdoc scientist at Harvard, trying to image biological events in his brain tissue study in 1955.2 By 1971, laser was induced as light source for the first time in confocal microscope.3‐4 In mid 1980s, confocal microscope has been evolved into its modern form, typically a beam scanning confocal microscope.5 A conceptual figure of confocal microscopy is shown in figure 2.1. The central idea of confocal microscope is to use two pinhole apertures at both ends of the optical path: one aperture is put in front of the laser light source, and another aperture is set in front of the photon detector module. Once these two pinhole apertures form optical conjugate plane, fluorescence signals from unwanted part of specimen which are out of focus can be blocked.6 In traditional wide field fluorescence microscope, the incident light excites the whole sample specimen. As a result, it is difficult to use such illuminating method to pinpoint a specific small volume of specimen, since fluorescence emitted from all parts of the specimen always arrive through the same optical path to the photon detector 26 simultaneously, forming unwanted background noise, lowering down signal-noise ratio. By using the confocal illuminating approach, we can block out lights from all parts other than the focal volume on the specimen, only allowing emission light from a very close region around the focal point on sample place to be detected and hence to achieve a high spatial resolution. In practical, the best vertical imaging resolution of confocal microscope can be 0.5µm, while the best horizontal imaging resolution can be 0.2 µm, making the spatial imaging volume close to the diffraction limit. In experiments, two additional points are noteworthy. Because of the fluorescence signal from out-of-focus part on the specimen will be blocked by the conjugate pinhole apertures, the overall signal will only be fluorescence from the focused volume in the specimen. Hence a low signal level is expected, although with a higher signal-noise ratio compared to traditional wide field microscopy. To have a practical detection, we will need photomultiplier tube (PMT) or Avalanche Photodiode (APD) as detector module. We use APD in our confocal microscopy setup as detector. Another point worth mentioning is that since the imaging capability of confocal microscope is limited in the focal volume of the specimen, it will require a scanning process to have an image of a large area of the specimen, i.e. a 5µm × 5µm region. For one single protein molecule having a few hundred of amino acid residues, such as 6-hydroxymethyl-7,8dihydropterin pyrophosphokinase (HPPK) or horseradish peroxidase (HRP) which has been used in our experiment, the overall size of the molecule is limited within 10 nm. Hence it will be an ideal imaging approach using confocal microscopy to detect protein molecules. In our studies, we selectively labeled two dye molecules Cy3 and Cy5 on a 227 singlee HPPK prottein moleculle while usin ng confocal m microscopy to study connformationall dynam mics of HPP PK moleculee, which willl be discusseed in Chapterr 3. Figure 2.1. A concep ptual figure of o confocal m microscopy.. Green liness indicate incident laser sou urce light, wh hile red lines stand for eemission fluoorescent lighht after excitaation of sam mple specimeen. 28 2.1.2 Principles of Forster Energy Transfer (FRET) Förster resonance energy transfer (FRET) spectroscopy, or fluorescence resonance energy transfer spectroscopy as an alternative term sometimes, has been regarded as one of the remarkable cornerstones in biophysical study field. The concept of FRET originates around 1948, named by Theodor Förster, a German Physical Chemist.7 The central idea is outlined below. When two different chromophore molecules are close to each other in a few tens of angstrom and getting excited by incident light, excitation energy can be transferred between the two dye molecules by dipole-dipole resonance interactions, as shown in figure 2.2. In this energy transfer, the chromophore sending out energy is named donor, and the chromophore that receiving energy is named acceptor. When a donor molecule is excited from its ground state (S0 in figure 2.1) to its excited state (S1 in figure 2.1) by incident light, the excited donor will disperse its energy in two processes that competing with each other: coming back to its ground state while emitting photon to form ‘fluorescence from donor’, or nonradioactively transfer this energy to the acceptor molecule, forming ‘fluorescence from acceptor’ by long-range inter molecular dipole-dipole coupling, which is named Förster energy transfer. The rate of Förster energy transfer can be calculated by equation 2.1.8 kT = 1 τD ×( R 6 ) R0 (2.1) In equation 2.1, kT is the energy transition rate constant, τD is the fluorescence lifetime of donor chromophore, R is the distance between donor molecule and acceptor molecule, R0 is a distance parameter named Förster distance, which depending on various factors: specific spectroscopic and mutual orientations of different chromophore 29 molecules, refractive index of solvent, and the overlap of emission spectrum of donor and absorption spectrum of acceptor.9 The parameter we observe in experiment is the quantum yield of Förster energy transfer, named FRET efficiency, described by equation 2.2. E= (Energy transferred from Donor to Acceptor)/(Energy absorbed by donor) (2.2) The relationship of FRET efficiency E and FRET rate constant kT can be described as equation 2.3. E= k ET k f + k ET + ∑ ki (2.3) In equation 2.3, kET is the FRET rate constant; kf stands for the radioactive decay rate of donor, while ki indicates all other processes in which donor molecule decay from its excited state. 330 Figurre 2.2. A con nceptual figu ure of the prrinciple of reesonance eneergy transferr between two different d chro omophore molecules. m Experimeental measureement of thee FRET efficciency reliess on the relattionship show wn in equation 2.4. The directly d obseerved parameeters are steaady state fluuorescence intensity of donorr (FD) and accceptor (FA). FDA indicattes the fluoreescence intensity of donorr with the ex xistence of acceptor, a whiile FD indicaates the fluorrescence inteensity of donorr when only donor moleecules exist in sample sollution. E= FD − FDA FD (2.4 4) 31 It has been discovered that the rate constant of such energy transfer is proportional to the inverse sixth power of the distance between donor and acceptor, as shown in equation 2.5. E FRET = 1 1 + ( r / R0 ) 6 (2.5) In equation 2.5, r is the distance between donor molecule and acceptor molecule, R0 is the Förster distance of the donor-acceptor pair. Hence, Förster resonance energy transfer efficiency can be an indicator of the distance of two different chromophore molecules, as long as the distance between the two chromophore molecules is in the scale of a few nanometers. FRET spectroscopy takes advantage of this phenomenon, using two or even more different dye molecules as donor and acceptor, labeling those dye molecules on certain positions of biological specimen, to monitor the distance between those positions, and hence to obtain structural information of biological specimen. The relationship between donor-acceptor distance and FRET efficiency make FRET spectroscopy become a ‘molecular scale ruler’ especially for biophysical studies. The effective distance of FRET spectroscopy is 10 to 100Å.10 On the other hand, when the distance between donor and acceptor is close to the Förster distance R0, FRET efficiency is most sensitive to the change of donor-acceptor distance, as shown in figure 2.3. Hence, in practical, FRET is widely used as a precise molecular level ruler to measure distance around 40Å to 60Å in biological specimens. 332 Figurre 2.3. The distance-FRE d ET efficienccy relationshhip. Successfu ul FRET meaasurement allso requires that approprriate chromoophore shoulld be ch hosen. The Förster F reson nance energy y transfer reqquires overlaap between eemission specttrum of dono or and absorp ption spectru um of accepttor, which m means that thhe energy releassed from don nor moleculees can be efffectively recceived by accceptor moleccules. In ouur studiees, we use Cyanine C 3 (C Cy3) as donorr and Cyaninne 5 (Cy5) aas acceptor, w while their specttrum profile is shown in figure 2.4. The T Forster distance of ddye pair Cy33 and Cy5 iss 5.4nm m. 11‐12 333 Figurre 2.4. Specttrum profile of Chromop phore moleccule Cy3 andd Cy5. 2.1.3 Principles of Total Intternal Reflection Microoscopy (TIR RFM) on is a comm mon physicaal phenomennon that happpening whenn Total inteernal reflectio h a boundaryy of differentt medium. S Since light iss a beaam propagatiing wave passses through also one o type of electromagn e et propagatin ng wave, tottal internal rreflection is iinvolved as a charaacter of lightt. In brief, on nce a beam of o light is paassing througgh a boundaary of two differrent optical medium, m wh hose refractiv ve index are n1 and n2 reespectively, w while n1>n2, partiaal of the ligh ht will go thrrough the meedium; partiaal of the lighht will be refflected back to thee same side of o the incideent light, as shown s in figu gure 2.5. However, whenn the angle formeed by incident light and the normal direction d to tthe boundaryy surface is larger than a certaiin angle, notted as θ in figure 2.5, thee incident ligght will be 100% reflecteed back 34 internally to the incidence side. This phenomenon is called total internal reflection, and the angle θ is named critical angle, as shown in figure 2.5. 13 One point need to be specially mentioned is that if a beam of light is shedding from on medium, hitting to another which has a lower refractive index, i.e., if n1<n2 in figure 2.5, total internal reflection phenomena will not occur in any angle. There is no concept as ‘critical angle’ anymore in this condition, and the term ‘total internal reflection’ does not refer to this situation. 14‐15 The physical principle of total internal phenomena can be explained by Snell’s law. When a beam of light is propagating from one medium with refractive index n1 to a different refractive index n2, the angles formed by the optical beam to the normal direction to the boundary surface are noted as θ1 and θ2 respectively, as shown in figure 2.5. Then the relationship between these two different refractive index and angles can be described by equation 2.6, which is called Snell’s law. n1 sin θ1 = n2 sin θ 2 (2.6) Hence, when n1 is larger than n2, sinθ1 will be smaller than sinθ2. That means a ‘bending’ of the light path, and a larger θ2 compared to θ1. As a result, when θ2 reaches 90o, θ1 is still at a sharp angle that between 0o and 90o. For any larger θ1 since then, the incident light will always be total internally reflected back to the original medium. The θ1 at that point is the critical angle, as shown in figure 2.5. 335 Figurre 2.5. A co onceptual fig gure of total internal refleection phenoomenon. There is an a interesting g and importtant side effeect of total innternal refleection pheno omenon, nam med evanesccent wave. When W a beam m of light is propagatingg in a medium m with refractive in ndex n1, hittin ng to the bou undary of annother mediuum with smaaller refracctive index n2 in an angle that is larg ger than criti cal angle annd hence youu get total intern nally reflecteed to the orig ginal medium m, there is sttill some waave ‘penetratting’ to the otherr medium, traavel along th he boundary of the two m mediums. A As shown in ffigure 2.6, evaneescent wave is a near-fieeld wave evaanescent wavve traveling along the x direction, and decay d along z direction. 36 The physical principle of evanescent wave is as below. First, after a beam of planar wave is hitting a boundary of two different medium, the transmitted part of the wave can be described by equation 2.7. k T = kT sin(θT )x + kT cos(θT )z (2.7) In equation 2.7, kT is the wave vector of transmitted wave, θT is the virtual angle of the transmitted wave to the normal direction of the boundary, x and z is direction unit vector shown in figure 2.6. From equation 2.6 we have sin(θT ) = n1 cos(θ1 ) n2 (2.8) The angle θ1 is the angle between incident wave and normal direction of the boundary surface, as shown in figure 2.6. Hence, when n1>n2, sinθT is larger than 1, resulting in a complex value cosθT: cos(θT ) = i sin 2 (θT ) − 1 (2.9) On the other hand, from electrodynamics, we have description of transmitted plane wave as below: E T = E 0 e i ( k T •r −ω t ) (2.10) Combined equation 2.7, 2.9 and 2.10, we have ET = E0e − kT sin 2 (θT ) −1z i ( kT sin(θT ) x −ωt ) e (2.11) 337 Equation 2.11 directly y shows the character off evanescent wave: traveeling throughh x direection, decay y along z dirrection. In brief, b evanesccent wave w will only be iintense withiin one th hird of the wavelength w starting s from m the boundaary surface. In our experriments, we use 532 nm lasers as excitatio on incident light, l hence tthe generateed evanescennt wave is nce approxim mately 170 nnm starting frrom the consiidered to be effective witthin a distan samp ple plane. Figurre 2.6. A co onceptual sch heme of evaanescent wavve when totaal internal refflection of optical beam occu urs. 38 An alternative representation of equation 2.10 is as below: 16 z I = I 0 exp(− ) d (2.11) In equation 2.11, I is the intensity of evanescent wave, z is the perpendicular distance to the sample surface, d is characteristic exponential decay depth, which can be described as below: d= 1 − λ sin 2 θ I ( 2 − 1) 2 4π n2 sin θ (2.12) In equation 2.12, λ is wavelength of incident light, n2 is refractive index of the transmitted substrate shown as figure 2.6; θ is the critical angle, while θI is incident angle which is the angle formed by incident light and normal direction to the sample plane. Total internal reflection microscope (TIRFM) takes advantage of evanescent wave to form a molecular-level fluorescence signal detection method. A conceptual graph of TIRFM is shown in figure 2.7. A beam of incident lasers is hitting through objective to the top surface of glass coverslip, and get entirely reflected back by tuning incident angle larger than critical angle. Evanescent wave will be generated close to the top surface of the coverslip, where the specimen is put. Immersed oil is always used to ensure that refractive index value is identical during the optical path of incident light before it hits the top surface of glass coverslip. Details of TIRFM in my experiments will be discussed in the experiment setup section. 339 Figurre 2.7. Concceptual scheeme of total internal i refleection microoscopy (TIRF FM). Greenn arrow w lines indicaate incident light source, while red liines indicatee signal beam m light emittted by samplle specimen after being excited e by evvanescent w wave. 40 2.1.4 Signal Detection techniques: Introduction to APD and EMCCD Introduction to Avalanche Photodiode (APD) In the magnetic tweezers-confocal FRET measurement which is the experiment performed in chapter III, we use avalanche photodiode (APD) as optical signal detection approach. In the magnetic tweezers-TIRFM measurement which is the experiment performed in chapter IV, we use electron multiplied charge coupled device (EMCCD) as optical signal detection method. In this section, we will briefly introduce the basic principles of these two fluorescence signal detection techniques. Avalanche photodiode (APD) is a type of semiconductor electronic device that based on photoelectric effect, converting light to electric signals with high sensitivity. The working principles of semiconductor photodiode as photodetector is shown in figure 2.8A and 2.8B. The core part is a PIN diode, which has one a p-type semiconductor and one n-type semiconductor at two ends, with a wide semiconductor region in middle. The p-type part and n-type part are usually heavily doped while the semiconductor region is undoped. When one photon comes in as external light signal, an electron with proper excited and raised from the valance band to the conduction band to generate an electronhole pair will be formed in the semiconductor region. External bias voltage will let these two electrons to drift quickly away from the junction region. Once there are light hitting the detector with multiple photons, such electron movement will form a current flow whose intensity is proportional to the incident light. In this way, light signals can be converted to electronic signals to be detected precisely and quantitatively. 41 However, in traditional PIN diode, only one electron-hole pair will be triggered by one incoming photon. For single molecule experiments, the light signal will always be weak, which means there may not to be enough photons to trigger electric signals with satisfied signal-noise ratio. In some single molecule experiments, there are even photons arriving to the detector one by one.17 APD will be used instead to discern signals when only a few photons are collected. As shown in figure 2.8C, the core part of APD is the built-in signal amplification region named depletion layer, typically a silicon photodiode structure, where electron multiplication occurs. When one photon arrived from the n-side, an electron-hole pair is formed. The n-side is negatively doped, while the p-side is positively doped, and a thin p-layer is coated on the n-side. An external reverse bias voltage is applied, which means p-side is connected to cathode, and n-side is connected to anode. The electron-hole pair in the depletion layer will then move towards respectively to the PN junctions due to external reverse bias voltage: electron will run back toward original n-side, and the ‘hole’ runs toward p-side. Once the external reverse bias voltage is high enough, i.e. higher than 105 V/m, the electron will collide to the thin p-layer on the n-side, resulting in ionization generating more electron-hole pairs. In this way, one single incident photon is capable to trigger multiple electron-hole pairs. Such process is named impact ionization, or avalanche effect. As a result of avalanche effect, the signal level become detectable, named current gain effect. The gain will be around 100 when reverse bias voltage is around 100-200 V. By appropriate doping techniques, the gain can even be high as 1000. 442 Figurre 2.8. Princciple of Phottodiode Mod dule used in oour experim ment. (A) Traaditional PIN N photo odiode when n there is no incident ligh ht. (B) Workking mechannism of tradittional PIN photo odiode when n light is inco oming. (C) A conceptuaal figure of aavalanche phhotodiode configuration. 43 Introduction to Electron Multiplied Charged Coupled Device (EMCCD) In the magnetic tweezers-TIRFM measurement which is the experiment performed in chapter IV, we use electron multiplied charge coupled device (EMCCD) to monitor fluorescence signal of released products from single molecule enzyme catalysis events. The core part of traditional CCD is a photoactive layer consisting of a silicon capacitor matrix array to sense photons from external light. Nowadays it is a common technique used in digital cameras or optical scanners. For single molecule level signal detection, similar to traditional photodiode, traditional CCD also cannot provide sufficient signal-noise ratio due to lower photon level in experiments. Hence, EMCCD is used instead. The principle of EMCCD is as described in figure 2.9. In brief, EMCCD also utilized the phenomenon of impact ionization to amplify the electron signal from active pixels hit by incident light. The core part of EMCCD is a gain register placed between the output amplifier and the shift register, as shown in figure 2.9. The gain probability at every stage in the gain register is typically small as 2%. However, in one EMCCD, there will be hundreds of stages in the gain register, and resulting in overall high gain. When an incoming image signal hits on the active pixels, the information of the image will be stored as converted electron signal in the image storage area. Then, the stored information can be either read out row by row from the serial shift register to generate a normal CCD image, or to pass through the electron multiply register, amplify signal via impact ionization, to generate an electron multiplied image. One point needs to be mentioned is that time for signal readout process in both CCD image and EMCCD 444 image cannot be neglected. It I may requirres tens of m millisecond ttime for one frame of image which con nsisting of 51 12 times 512 2 pixels. Figurre 2.9. Princciple of Electtron Multipllied Charge C Coupled Devvice (EMCC CD). 45 2.1.5 Basics of Magnetic Tweezers: Force Calibration An introduction of principle, history and application of single molecule magnetic tweezers has already been discussed in chapter 1.4. In this section, we will go through some experimental details of magnetic tweezers. In practical, when setting an experiment using magnetic tweezers as single molecule level force manipulation approach, two major aspects need to be deliberately considered. First of all, an appropriate observation approach needs to be correlated with magnetic tweezers. This is because unlike other single molecule level manipulation technique such as AFM or optical tweezers, magnetic tweezers does not have a way to directly obtain the sample conformation by its own. In physical essential aspect, the core part of magnetic tweezers will be a piece of magnet, despite sometimes it can be designed into many different complicated forms. Hence, to carry out a single molecule level magnetic tweezers experiment, at least one type of single molecule optical observation setup needs to be set to provide information of target sample molecules during the experiment. Secondly, appropriate combination of magnetic field and magnetic beads need to be chosen to convey the magnetic force onto target sample molecules. In this section, our discussion will focused on force calibration for magnetic tweezers. And details of correlating magnetic tweezers setup with correlated single molecule optical observation methods will be discussed in section 2.2. Mechanical force from external magnetic field is applied on a targeted protein through a paramagnetic bead linked covalently to the single protein molecule. To quantitatively understand the force that applied on protein molecules by the magnetic 46 tweezers, we note that there are a number of specific approaches to estimate the mechanical forces applied though a magnetic field on a paramagnetic bead: (1) Measuring and model analyzing the Brownian motions of a tethered paramagnetic bead;18 (2) Monitoring the dragging motion of a small number of magnetic beads in liquid environment with known viscosity;19,20 (3) Observing the displacement of a micropipette with a magnetic bead attached at its end, etc.19 Different methods for measuring torque on magnetic beads have also been developed.21 Nevertheless, each of the estimation approaches bears specific merit of estimation with certain error bars. We have applied a model analysis based on the measured magnetic field strength curve (Figure 2.8) as a function of the distance between the magnetic tip and the sample surface. We calibrate the applied force by estimating the magnetic field gradient to get the magnetic moment of the beads tethered on the single protein molecule. For a magnetic bead in an externally-produced magnetic field B, noting its magnetic moment as m, then the potential energy U is: (2.13) U = -m × B For a given magnetic bead, its magnetic moment m is the product of the volume magnetization M and volume V of the bead. Therefore, the force F that is applied on the magnetic bead can be calculated: F = -∇U = -∇(-m ⋅ B ) = m ⋅∇B = MV ⋅∇B = MV ∂B ∂z (2.14) In our experiments, the magnetic field applied is approximately 1100 Gauss. As an approximation, we only consider the magnetic field gradient in one direction 47 perpendicular to the sample plane. Thus the field gradient can be estimated from the curve shown in Figure 2.10. In this way the value of field gradient is calculated to be 55±15 T/m. When calculating the field gradient, position error of up to 1mm is taken into consideration as uncertainty of distance between the magnet and the sample plane. The volume V of paramagnetic bead is 0.6×10-18m3, and the volume magnetization M is 43×103 A/m.22 In our calculation, we have considered the factor that M here is the saturation magnetization, an approximation that may bring error less than 25%. Hence the force is calculated 1.4±0.4 pN from equation 2.14. The typical force applied to the targeted single-molecule HPPK proteins is roughly 1-3 pico-Newton that is weaker than a typical hydrogen bonding force of 6-9 pico-Newton. Figure 2.10. Magnetic field-Distance curve of the magnet used in experiment. Each data point is repetitively measured for five times. The blue point indicates the position of the magnet in the experiment which is set 4 mm above the sample plane to generate a magnetic field with approximately 1100 Gauss strength. 48 2.2 Experimental Details 2.2.1 Experimental Setup of Single Molecule FRET Correlated with Magnetic Tweezers In chapter III, the measurement was carried out by a correlated setup combining a two-channel laser scanning microscope with magnetic tweezers, to take single-molecule FRET imaging as simultaneous optical observation of protein conformation while mechanical manipulation of the same protein molecule by magnetic tweezers is performing. The setup configuration is shown in figure 2.11. In brief, we use the singlemolecule photon stamping approach to record the single- molecule FRET fluctuation time trajectories photon by photon for both the donor and acceptor simultaneously. The fluorescence images and photon-counting trajectories are acquired with an inverted confocal microscope (Axiovert 200, Zeiss). The excitation laser (532 nm continuouswave (CW) Crystal laser) confocal beam is reflected by a dichroic beam splitter (z532rdc, Chroma Technology) and focused by a high-numerical aperture objective (1.3 NA, 100×, Zeiss) on the sample at a diffraction limited spot of ~300 nm diameter. In order to obtain the fluorescence images and intensity trajectories, the emission signal is split by using a dichroic beam splitter (640dcxr) into two color beams c entered at 570 nm and 670 nm representing Cy3 and Cy5 emissions, respectively. The signals from two channels are detected by a pair of Si avalanche photodiode single photon counting modules (SPCMAQR-16, Perkin Elmer Optoelectronics). Typical images (10 μ m × 10 μ m) are acquired by continuously raster-scanning the sample over the laser focus with a scanning speed of 449 4ms/p pixel, with each e image of o 100 pixelss × 100 pixells. The fluorescence inttensity trajecctories of thee donor (Cy 3) and accep pter (Cy5) arre recorded bby a two-chaannel Picoh harp 300 (PiccoQuant) ph hoton-stampiing set-up. A permanentt magnet is cconnected onn an ind dependent z-axis stage to control its distance to the sample gglass coversllip, while X-Y dirrection in-plaane movemeent is controllled by the tuuning the tw wo-layer sam mple stage byy comp puter. Figurre 2.11. Exp perimental setup of single molecule FRET-Magnnetic Tweezzers specttroscopy. 550 2.2.2 Experimental Setup off Single Mole lecule TIRFM FM Correlateed with Maggnetic Tweeezers In chapterr IV, the meaasurement was w carried oout by a correelated setup combining a inverrted laser scaanning micro oscope with magnetic tw weezers, to taake single-m molecule TIRF F imaging as simultaneou us optical ob bservation off enzymatic activity whiile mech hanical manipulation of the t same enzzyme molecuule by magnnetic tweezerrs is perfo orming. The setup p configuration is shown in figure 2.12. In brief, we carried oout the TIRF F measurements by y using an in nverted confo ocal microsccope (Olymppus IX 71 wiith 60 x W laser (Crysstalaser) gennerating evannescent wavee for total objecctive) with a 532 nm CW intern nal excitation n. Emitted signal s are filltered with oone beam spllitterfilter (C Chroma Techn nology, z532 2rdc), one 54 45 nm long--pass filter annd then beinng collected bby an Electtron Multiply ying Charge Coupled Deevice (EMCC CD: ProEM 512B, PI coo.). 51 Figure 2.12. Experimental setup of single molecule TIRF-Magnetic Tweezers spectroscopy. 2.2.3 Steered Molecular Dynamics (SMD) Simulation In chapter V, we performed steered molecular dynamics (SMD) simulation to have a single molecule level understanding of the impact of conformational manipulation to the function of target protein molecule. The scene at molecule level in our studies is as below. Firstly, we immobilized one single enzyme protein molecule at one given residue position. A external pulling force is then applied to another given residue position on the sample enzyme protein molecule. In chapter III, we achieved conformational manipulation of HPPK protein molecule by magnetic tweezers at single molecule level. In chapter IV, we studied HRP enzyme protein using the same conformational manipulation approach to discover what impact such conformational manipulation can have to protein function. Hence, Steered MD simulation can provide us additional valuable information of a molecule level scene connecting the studies in chapter III and chapter IV. There are several different software packages for the purpose of normal MD simulation, such as GROMACS, CHARMM, AMBER, etc. However, the scene in our studies that ‘pulling one single protein from two given residue position’ requires a special type MD simulation named Steered MD (SMD), with NAMD as software package respectively. NAMD was developed by the Theoretical and Computational Biophysics Group in the Beckman Institute for Advanced Science and Technology at the University of Illinois at Urbana-Champaign. More details of our SMD simulation will be discussed in chapter V. 52 2.3 Materials and Sample Preparation In our experiments, we prepared single molecule immobilized protein samples in the way described in Figure 2.13.In brief, we tether a single protein molecule to a modified glass coverslip at one end by TESPA-DMS linkers and bound to a superparamagnetic bead (Dynabeads® MyOne™ Streptavidin T1, 1.05-µm diameter, Invitrogen Company) at the other end via biotin-streptavidin linkers. The glass coverslip is modified as below. Firstly, a clean glass coverslip is immersed overnight in NaOHethanol solution. The coverslip was next washed by distilled water, blow-dried by air flow, and incubated with a DMSO solution containing a mixture in 10% concentration consists of TESPA and isobutyltrimethoxysilane in 1:10000 ratio overnight. The low concentration of each solution was to make sure that the distribution of protein molecules on cover glass is separate so that one bead does not attach to multiple protein molecules. More details such as incubation time and buffer pH will be discussed in chapter III and chapter IV specifically. 553 Figurre 2.13. A conceptual scheme s of siingle molecuule protein im mmobilizatioon method. A cleean cover glaass was treatted by 3-amiinopropyltrieethoxy-silanne (TESPA) m mixed in isobu utyltrimethox xysilane in 1:10000 1 ratio o in DMSO ssolution. Prrotein molecuules are tetherred onto the siliconized cover glass at a one end bby Dimethyl suberimidatte-2HCl (DMS S-2HCl) which reacting with amine group on TE ESPA, and aat the other eend to a param magnetic beaad by biotin--streptavidin n bonding. P Protein molecules use thee amine group p from its Ly ysine residuees to form bo ond at both eends. As a rresult, all Lyysine residuees are acccessible to become the linking posiition of one ssingle proteiin molecule, leading to a multiiple-possibility of protein n tethering condition. c D Details will bbe discussedd in chapter III an nd chapter IV V. 54 2.4. Selection of Magnetic Beads We chose the magnetic beads with 1 µm diameter for our experiment in order to apply force at pN scale on protein molecules. In experiment, an easy way to have a quick test of the applied force on magnetic beads is using the Stokes Formula Force=6πƞrv, where ƞ is the viscosity of the solution; r is radius of the magnetic bead; and v is the moving speed through the solution monitored from microscope. Although we use a different approach to quantitatively estimate the applied force by our magnetic tweezers, as shown in our previous publication, we still rely on watching velocities of free magnetic beads in solution to have an estimation of the force scale we applied. We assume our PBS buffer solution has similar viscosity as water, since the concentration of the buffer solution is low as 50mM. The diameter of eyepiece on microscope is approximately 3 cm, while the objective amplification is 60 and eyepiece amplification is 10. As a result, by observing through eyepiece we can monitor an area of 50 µm. As shown in figure S5, we can see that magnetic beads with small diameter at 100 nm scale need to be driven faster than 1 mm/s to generate even 1 pN force, indicating that it is not a good choice to use beads at 100 nm scale for our experiment. On the other hand, although based the Stokes Formula, larger beads always appears to be better, we also need to consider that our experiment is observing protein molecules, with their size at 1 to 10 nm scale. Hence, we hope to limit the bead size as small as possible to decrease the impact of the magnetic beads to local solution environment. As a compromise of these two thoughts, we chose the 1µm size beads for our measurements. 55 Fig. 2.14. Force-velocity response of magnetic beads in different size. 56 2.5 References 1 Goldmann, H. Spaltlampenphotographie und –Photometrie. Ophthalmologica 1939, 98 (5/6): 257–270. 2 Minsky, M. Microscopy Apparatus: US 3,013,467. 3 Egger, M. D. (1971). Scanning Laser Microscope for Biological Investigations. Applied optics. 1971, 10 (7): 1615–1619. 4 Amos, W.B.; White, J.G.: How the Confocal Laser Scanning Microscope Entered Biological Research. In: Biology of the Cell / under the Auspices of the European Cell Biology Organization. Band 95, Nummer 6, September 2003, S. 335–342. 5 Prasad, V.; Semwogerere, D.; Weeks, E.R. Confocal Microscopy of Colloids. J. Phys.: Cond. Mat. 2007 19, 113102. 6 Förster, T. Zwischenmolekulare Energiewanderung und Fluoreszenz. Annalen der Physik 1948, 437, 55-75. 7 Förster T. Fluorescence of Organic Compounds Gettingen: Vandenhoeck & Ruprecht: 1951:312. 8 Stryer, L.; Haugland, R. P. Energy Transfer: A Spectroscopic Ruler. Proc. Natl. Acad. Sci. USA 1967, 58, 719-730. 9 Clegg, R.M. Fluorescence Resonance Energy Transfer. Curr. Opin. in Biotech. 1995, 6:103-l 10. 10 Ha, T.; Enderle, T.H.; Ogletree, D.F.; Chemla, D.S.; Selvein, P.R.; Weiss, S. Probing the Interaction Between Two Single Molecules: Fluorescence Resonance Energy Transfer Between a Single Donor and a Single Acceptor. Proc. Natl. Acad. Sci. USA 1996, 93, 62646268. 57 11 Deniz, A.A.; Dahan, M.; Grunwell, J.R.; Ha, T.; Faulhaber, A.E.; Chemla, D.S.; Weiss, S.; Schultz, P.G. Single-Pair Fluorescence Resonance Energy Transfer on Freely Diffusing Molecules: Observation of Förster Distance Dependence and Subpopulations Proc. Natl. Acad. Sci. USA 1999, 96, 3670–3675. 12 Ambrose, E.J. A Surface Contact Microscope for the Study of Cell Movements. Nature 1956, 178 (4543): 1194. 13 Yanagida, T.; Sako, Y.; Minoghchi, S. Single-Molecule Imaging of EGFR Signalling on the Surface of Living Cells. Nature Cell Biology 2000, 2 (3): 168–172. 14 Axelrod, D. Total Internal Reflection Fluorescence Microscopy in Cell Biology". Traffic 2001, 2 (11): 764–774. 15 Axelrod, D. Cell-Substrate Contacts Illuminated by Total Internal Reflection Fluorescence. J. Cell Biol. 1981, 89, 141-145. 16 He, Y.; Lu, M.; Lu, H.P. Single-Molecule Photon Stamping FRET Spectroscopy Study of Enzymatic Conformational Dynamics. Phys. Chem. Chem. Phys., 2013, 15, 770-775. 17 Smith, S. B.; Finzi, L.; Bustamante, C. Direct Mechanical Measurements of the Elasticity of Single DNA Molecules by Using Magnetic Beads. Science 1992, 258, 1122-1126. 18 Haber, C.; Wirtz, D. Magnetic Tweezers for DNA Micromanipulation. Rev. Sci Instrum. 2000, 71, 4561-4570. 19 Kollmannsberger, P.; Fabry, B. High-Force Magnetic Tweezers with Force Feedback for Biological Applications. Rev. Sci Instrum. 2007, 78, 114301. 20 Forth, D.; Sheinin, M.Y.; Inman, J.; Wang, M.D. Torque Measurement at the SingleMolecule Level. Annu. Rev. Biophys. 2013, 42, 583-604. 21 Note: the value is according to the product specification from Invitrogen Company. 58 CHAPTER III. MANIPULATING AND PROBING ENZYMATIC CONFORMATIONAL FLUCTUATIONS AND ENZYME-SUBSTRATE INTERACTIONS BY SINGLE-MOLECULE FRET-MAGNETIC TWEEZERS MICROSCOPY 3.1 Introduction Conformational change of protein molecules is often critical for the biological functions, affecting the affinity and selectivity of protein-protein and protein-ligand interactions, and further regulating the catalytic activity of enzymatic reactions.1-4 For example, an enzyme can have different activities with different conformations.5-7 Thus, manipulating protein conformations can be an effective approach to study the relationship between protein conformation and function.8-30 One of the central questions in protein functions is the impact of ligand binding to conformational fluctuation or conformational flexibility changes of protein molecules, especially enzyme-substrate interaction.3 The answer to this question serves a critical understanding of the enzyme-substrate interactions and the enzymatic active transition state formation. In recent years, a number of novel single-molecule approaches combining single-molecule optical spectroscopy with mechanical force manipulation approaches have been developed to achieve protein conformational manipulation, such as atomic force microscope (AFM), optical tweezers, and magnetic tweezers, etc.9-11,21-36 Here we report our newly developed approach using magnetic tweezers correlated with single-molecule FRET spectroscopy to study ligand-binding impact on enzymatic conformation by force manipulating single enzyme molecule conformation with 59 simultaneous optical observation of the enzyme conformational fluctuations under different conditions of with and without enzymatic substrate. Compared with other approaches for manipulating single protein molecules, such as AFM or optical tweezers, magnetic tweezers has a number of desirable and complimentary specificities: (1) magnetic tweezers can apply a pulling force either in a fine scale as small as sub-picoNewton37 or in a relative large scale close to nanoNewtons;38 (2) magnetic tweezers does not require physical contact or chemical contact to target molecules; (3) magnetic tweezers does not induce either photo-damage to the sample or a photon background noise to a correlated simultaneous single-molecule spectroscopic measurement; (4) magnetic tweezers allows manipulating conformation of a large number of molecules simultaneously as long as the molecules are tethered to paramagnetic micro beads. These specificities make the magnetic tweezers approach promising for protein conformational manipulation. Since 1990s, extensive studies on manipulating single biological molecules by using magnetic tweezers have been reported.39-45 The applications of magnetic tweezers manipulating biological molecules have been extended from DNA wringing46,47 to polymer protein molecules pulling48, 49. The correlated theoretical simulations have also been developed in recent years.48,49 Nevertheless, to our knowledge, conformational manipulation by magnetic tweezers and correlated simultaneous single-molecule FRET spectroscopic analysis of a single protein molecule has not been reported. 60 3.2 Materials and Methods 3.2.1 HPPK Protein In our experiment, we chose fluorescence dye labeled 6-hydroxymethyl-7,8dihydropterin pyrophospho- kinase (HPPK) as a model system to measure the FRET and magnetic tweezers manipulations in the solutions with and without enzymatic substrates. HPPK is an 18 kDa 158-residue monomeric enzyme protein molecule with the biological function to catalyze the transferring of pyrophosphate from ATP to 6-hydroxymethyl-7,8dihydropterin (HP), releasing adenosine monophosphate (AMP) and 6-hydroxymethyl7,8-dihydropterin pyrophosphate (HPPP) as products.50-53 The fluorescent dyes Cy3 (donor)/Cy5 (acceptor) was labeled to the mutated amino acid residue 48 on loop 2 and residue 151 close to the active site of the enzyme, respectively, as shown in Figure 3.1. HPPK molecules were bound to the glass cover slip at one end by 3triethoxysilylpropylamine-Dimethyl Suberimidate links and attached to a superparamagnetic bead at the other end by biotin-streptavidin links. The HPPK molecule was labeled at residue position 48-151 with Cy3 and Cy5 dye molecules respectively. We choose Cy3-Cy5 dye labeled HPPK as a model system to study the effect of external force triggering on enzymatic conformational dynamics by using combined magnetic tweezers manipulations and correlated FRET measurement in the solution with and without enzymatic substrates. 61 Figure 3.1. Structuraal informatio on of HPPK molecule. (A A) Crystal sttructure of H HPPK moleccule from protein databan nk (PDB ID: 1HKA). Th he mutated dyye-labeled reesidue and L Lysine residuue has been pointed out (black spotss on the molecule). The HPPK moleecule was labbeled at resiidue 4 with h Cy3 and Cyy5 dye moleccules which are the greeen and red sppot in the schheme position 48-151 respectiv vely. (B) An example of single moleccular image of the HPPK K molecule oobserved by confocal microscope. (C) Full seequence of HPPK H proteinn. 62 3.2.2 Sample Preparation As shown in Figure 3.1, the fluorescent dyes, Cy3 and Cy5 as FRET donor and acceptor, were labeled to the mutated amino acid residue 48 on loop 2 and residue 151 close to the active site of the enzyme, respectively. The Cy3/Cy5 fluorescent dye pair was statistically labeled to the mutated enzyme with thiolation. HPPK molecules with this type of dye labelling have been used to study conformational dynamics in previous published work from our group.54 The HPPK molecules were bound to the glass cover slip at one end by 3-aminopropyltriethoxy-silane (TESPA)-Dimethyl Suberimidate•2HCl (DMS) linkers and linked to a super-paramagnetic bead (Dynabeads® MyOne™ Streptavidin T1, 1.05-µm diameter, Invitrogen Company) at the other end via biotinstreptavidin bond. Protein immobilization was carried out through a routine procedure shown in figure 3.2. In brief, a clean glass coverslip was immersed overnight in NaOHethanol solution. The coverslip was next washed by distilled water, blow-dried by air flow, and incubated with a DMSO solution containing a mixture in 10% concentration consisting of TESPA and isobutyltrimethoxysilane in 1:10000 ratio overnight. The coverslip was then washed by distilled water and consecutively transferred and incubated for 4 hours in each system below: 15 mL PBS buffer solution pH=8.0, containing 10nM Dimethyl Suberimidate•2HCl (DMS•2HCl); 15mL PBS buffer solution pH=7.4, containing 1nM HPPK; 15 mL PBS buffer solution pH=7.4, containing 10 nM NHSPEO12-biotin; 15 ml PBS solution pH=7.4, containing 1µl magnetic beads stock solution which is commercial available. The low concentration of each solution was to make sure that the distribution of protein molecules on cover glass is adequately separated so that one bead does not attach to multiple protein molecules. Meanwhile, low concentrations 63 of TESP PA are used to ensure that immobiilized protein moleculees are distriibuted separately enough from f each other o for obttaining sing gle moleculle FRET im mages. Figure 3.2. Preparatiion of singlee molecule HPPK H samplee. We tethereed protein m molecules at one end to the coverslip by b Dimethyll suberimidatte-2HCl (DM MS-2HCl) annd at the othher end to a 1 µm n-streptavidin n bonding. F Force was appplied by addding externaal size paramagnetic beead by biotin magneticc field and heence the mollecule could d feel it throuugh the beadds. In n our experiments, we conducted the single-m molecule FR RET measu urements wiith simultan neous magn netic tweezeers pulling of o HPPK en nzyme moleecules in PB BS buffer solution under the conditions c with w and wiithout the en nzymatic reeaction subsstrates. Thee me moleculees were imm mersed in a solution co ontaining 50 0 mM pH=7 7.4 immobillized enzym PBS bufffer solution n as imaging g buffer and d 1 mM 6-h hydroxy-2,5 5,7,8-tetram methylchrom man- 64 2-carboxylic (Trolox) solution as oxygen scavenger to protect dye molecules from photobleach. We also have noted that either biotin or DMS can only be tethered to a HPPK molecule via connection with lysine in the amino acid sequence, which leads to multiple possible tethered condition of the protein molecule to coverslip or magnetic beads. However, in each FRET measurement, we focused on a specific individual HPPK molecule during our repetitive manipulation by magnetic tweezers. Consequently, although we did not necessarily pinpoint that a pair of specific lysine residues tethered to a specific protein molecule, our observation of the reproducible FRET changes under periodically applied magnetic field demonstrates that successful single-molecule level protein conformational manipulation is achieved. All the attachment to HPPK molecule, either biotin or DMS, can only be fulfilled via connection with lysine in the amino acid sequence. Hence there are five possible positions in an HPPK molecule to allow attachment of magnetic particle via biotin or attachment to cover glass via DMS. As a result, there are five possible positions on one HPPK molecule available to attachment to either coverslip or magnetic bead. In the reaction two of these five positions will be occupied by attachment to either coverslip or magnetic bead. The combination brings multiple different possible types for the relative position of chromospheres and the lysine attached to glass or bead on a single HPPK molecule. Although from the crystal structure we can preclude some tethering conditions that are less possible: for example, in figure 3.1A we can see that it is essentially impossible for two linkers to consecutively tether on residue 154 and residue 157, we still are not able to pinpoint one deterministic specific amine residue pair for protein tethering. On the other hand, during our FRET measurement, we focused on one certain HPPK 65 moleculee, no matter what w certain n type it is. Iff we can obsserve its reprroducibly FR RET change under perriodically ap pplied magneetic field, wee are able to say conform mational mannipulation iss achieved d via magnetiic tweezers, although wee do not knoow which cerrtain two of the five lysinne on the HP PPK protein n is attached.1-3 3.2.3 Ex xperimentall System The T essentiaal componen nt of our maagnetic tweeezers devicce is a homeemade coneeshape peermanent magnet m moun nted on an independen i nt 3D translaational mov vement stag ge that conttrols the mo ovement of the magnett, as shown in Figure 3 3.3. Details of the setup p have alreeady been discussed d in n Chapter 2.2.1. Figure 3.3. 3 A conceptual scheeme of the experimenta e al system. (A A) Cy3-Cy y5 labeled HPPK kinase k moleccules are tethered on a modified g glass coversslip that waas positioned d in a buffer solution ch hamber. Th he inset paneel presents tthe conceptt of the con nformationaal 66 manipulation of a single HPPK molecule by magnetic tweezers. The cover glass is treated by 3-aminopropyltriethoxy-silane (TESPA) and isobutyltrimethoxysilane in 1:10000 ratio. Dimethyl Suberimidate•2HCl (DMS) is used as cross linker to immobilize HPPK protein molecule on the treated cover glass. The immobilized HPPK molecules are tethered through NHS-PEO12-biotinlinking the lysine residue of HPPK to the streptavidin-coated magnetic beads. (B) Magnetic Field-Distance curve of the magnet used in experiment. The blue data point indicates the position of the magnet in our experiments: the magnet is set 4 mm above the sample plane to generate a magnetic field with approximately 1100 Gauss. 3.2.4 Force Calibration In our experiments, mechanical force from external magnetic field is applied on a targeted protein through a paramagnetic bead linked covalently to the single protein molecule. As the discussion in chapter 2.1.5, we calibrate the applied force by estimating the magnetic field gradient to get the magnetic moment of the beads tethered on the single protein molecule. For a magnetic bead in an externally-produced magnetic field B, noting its magnetic moment as m, then the potential energy U is: U -m ⋅ B (3.1) For a given magnetic bead, its magnetic moment m is the product of the volume magnetization M and volume V of the bead. Therefore, the force F that is applied on the magnetic bead can be calculated: F = -∇U = -∇(-m ⋅ B ) = m ⋅∇B = MV ⋅∇B = MV ∂B ∂z (3.2) In our experiments, the magnetic field applied is approximately 1100 Gauss. As an approximation, we only consider the magnetic field gradient in one direction 67 perpendicular to the sample plane. Thus the field gradient can be estimated from the curve shown in Figure 3.3B. In this way the value of field gradient is calculated to be 55±15 T/m. When calculating the field gradient, position error that up to 1mm is taken into consideration as uncertainty of distance between the magnet and the sample plane. The volume V of paramagnetic bead is 0.6×10-18m3, and the volume magnetization M is 43×103 A/m.55 In our calculation, we have considered the factor that M here is the saturation magnetization, an approximation that may bring error less than 25%. Hence the force is calculated 1.4±0.4 pN from equation 3.2. The typical force applied to the targeted single-molecule HPPK proteins is roughly 1-3 pico-Newton that is weaker than a typical hydrogen bonding force of 6-9 pico-Newton. 3.3 Results and Discussion 3.3.1 FRET Measurement Figure 3.4A shows a pair of FRET donor-acceptor (D-A) fluorescence intensity trajectories from a single Cy3-Cy5 labeled HPPK molecule under force manipulation by magnetic tweezers. The FRET efficiency E is calculated from equation 3.3, in which ID and IA stand for the emission intensity of donor and acceptor, respectively. Figure 3.4C, the histogram of the FRET efficiency, shows the distribution of FRET efficiency. E=IA/(ID+IA) (3.3) The FRET efficiency reflects the distance between the two dyes labeled on protein molecules, described by equation 3.4, in which R is the distance between donor Cy3 and acceptor Cy5 while R0 is a constant determined by the transition donor-acceptor dipole– dipole interaction. In this experiment, when a pulling force is applied by the external 68 magneticc field, we are able to probe p the conformatio onal changess from the ssimultaneou us single-m molecule FR RET efficien ncy trajecto ories. EFRET=1/[1+(R/R0)6] (3.4) Figure 3.4. 3 Single-molecule FRET F data obtained o fro om a Cy3-C Cy5 labeled HPPK und der magneticc field man nipulation. (A) ( A portion of a pairr of single-m molecule flu uorescence intensity y time trajecctories of FR RET donor (green linee) and accep ptor (red lin ne). (B) The FRET effficiency caalculated fro om the pair of fluoresccence intenssity trajecto ories of the donor an nd acceptor in A. (C) The T FRET efficiency d distribution n deduced frrom B. 69 3.3.2. Repetitive Conformational Manipulation of Single HPPK Molecule Observed by FRET Spectroscopy Figure 3.5A shows the FRET efficiency distribution measured from a single HPPK enzyme under the enzymatic reaction conditions with the substrate of ATP and HP added in PBS buffer. With the magnetic field applied, the mean of the FRET efficiency is significantly shifted from 0.5 to 0.3, which suggests that the single-molecule HPPK enzyme is stretched out in conformation under the external pulling force. The result shown in Figure 3.5 demonstrates that our combined technical approach of magnetic tweezers correlated single-molecule FRET spectroscopy is sensitive and capable of manipulating and measuring molecule conformational changes simultaneously. To further demonstrate the reproducibility and effectiveness of the force manipulation of enzyme conformations by the magnetic tweezers correlated single-molecule FRET spectroscopy, we have conducted a repetitive force pulling and releasing manipulation of single HPPK enzyme molecules. Figure 3.5B shows that the single-molecule FRET efficiency toggles between 0.5 and 0.3 reflecting the enzyme conformational changes due to the manipulation by the external force pulling and releasing, demonstrating high reproducibility and feasibility of the force manipulation of the conformational changes of the single-molecule enzymes. It is intriguing that the FRET distribution shows a bimodal distribution pattern around efficiency value 0.2 when the enzyme molecule is pulled by magnetic force, which is most likely due to the force perturbation of the molecule, and the molecule still has significant conformational flexibility under the weak force manipulation. Nevertheless, the focus of this control experiment is to demonstrate the feasibility of the repetitive force manipulation of the overall enzyme conformational 70 changes and distributions whille the confo ormation flu uctuations o of the enzym me are still allowed and measurrable. Figure 3.5 3 Repetitiive force pu ulling and reeleasing maanipulation of individu ual kinase enzyme molecules. (A) The FRET F efficieency distrib butions of siingle HPPK K moleculess f pullin ng (Red) and releasing (Blue) man nipulation. (B) The FR RET efficiency under a force responsee of a singlee HPPK pro otein molecu ule being reepetitively ttoggled witth (Red) and d without (Blue) the external e maagnetic forcce. These F RET distrib butions are obtained frrom a series of o continuo ous single-m molecule FR RET measurrements witth the substtrate of ATP P and HP added a in the PBS buffe fer solution. 71 3.3.3. Probing Conformational Flexibility of Single HPPK Protein Molecule by Single Molecule FRET-Magnetic Tweezers Spectroscopy In an enzymatic reaction, the enzyme-substrate interaction is the crucial step determining the overall reaction dynamics as well as the reactivity and selectivity, according to the Michaelis-Menton mechanism and recent experimental and theoretical studies.56-66 The enzyme-substrate complex formation can regulate both static and dynamic conformations of the enzyme, and the enzyme-substrate complex requires a specific molecular conformation to form an active enzyme-substrate complex state ready to react and convert the substrate to the product. By probing the conformational fluctuations of single-molecule enzyme under the conditions of with substrate and without substrate, we have observed a significant change in conformational fluctuation distribution induced by the external force. Figure 3.6A and 3.6D show the enzymatic conformational distributions of HPPK in the buffer solution without the substrate, under the conditions of without (Figure 3.6A) and with (Figure 3.6D) the external pulling force, respectively. Figure 3.6B and 3.6E show the enzymatic conformational distributions in the buffer solution with the substrate of 100 μM ATP, 100 μM HP and under the conditions of without (Figure 3.6B) and with (Figure 3.6E) external pulling force manipulation, respectively. Comparing the distributions in Figure 3.6A and Figure 3.6B, measured under no external force manipulation, it is remarkable that the enzyme-substrate interaction narrows the conformational fluctuation range significantly, indicated by the smaller standard deviation of the FRET efficiency distribution (Figure 3.6B), which suggests that the enzyme-substrate interaction decreases the enzymatic conformational flexibility and the overall spatial accessibility. It is known that the 72 enzyme-substrate interaction can narrow and limit the enzyme conformational flexibility and accessible space, according the well demonstrated conformational selection mechanism or induced fit mechanism.67-73 However, due to the external force manipulations, the enzyme-substrate interaction is not able to cause a significant change in standard deviation of conformational fluctuation distributions, as shown in Figure 3.6D and Figure 3.6E, suggesting the external force manipulation provides a dominating impact on the enzyme conformational flexibility and limits the impact of the enzyme-substrate interaction on the enzyme conformational flexibility. For more quantitative understanding of the impact of enzyme-substrate interaction on enzymatic conformation fluctuation and the impact of external force manipulation on the enzyme-substrate interaction, we further use the standard deviation of FRET efficiency distribution to quantitatively characterize the broadness of the FRET efficiency distributions as well as the conformational flexibility.74,75 More flexible enzymatic conformation gives a wider enzymatic conformational fluctuation distribution in range, and a larger standard deviation of the conformational distribution. The results (Figure 3.6C and 3.6F) suggest that (1) the enzyme molecules without substrate have more flexible conformational fluctuations, which is indicated by the larger standard deviation in the FRET efficiency distribution; (2) the enzyme molecules with substrate interaction have more spatially confined conformational changes and less flexible conformational fluctuations, which is indicated by the smaller standard deviation in the FRET efficiency distribution; and (3) an external force pulling on an enzyme molecule decreases the impact of selective binding-folding enzyme-substrate interaction at the enzymatic active site. This attribution is further supported by the results measured under the conditions 73 with and d without su ubstrate presence: the enzymatic conformational distrib butions undeer a pulling force f perturrbation (Fig gure 3.6D, 3.6E, 3 and 3. 6F) show leess differen nce in the standard d deviation in i FRET effficiency com mparing to the same sttandard dev viation measureed in HPPK without thee pulling fo orce perturb bation (Figu ure 3.6A, 3.6 6B, and 3.6 6C) . Figure 3.6. 3 Perturb bing and chaaracterizing g enzyme-su ubstrate bin nding interaaction by sin nglemoleculee FRET maagnetic tweeezers micro oscopy. (A)) Distributio on of FRET T efficiency y of a single ap po-HPPK molecule. m (B B) Distribution of FRE ET efficiency y of HPPK in ATP and d HP substratee solution. (C) The sttandard dev viation of FR RET efficieency of sing gle HPPK moleculees measured d under the conditions of with (so olid line) an nd without ((dashed linee) 74 substrate in buffer solution and without the force perturbation. (D) Distribution of FRET efficiency of a single HPPK molecule under magnetic force pulling and without substrate ATP and HP added. (E) Distribution of FRET efficiency of a single HPPK molecule under magnetic force pulling in the solution with substrate ATP and HP added. (F) The standard deviation of FRET efficiency of single HPPK molecules measured under the conditions of with (solid line) and without (dashed line) substrate in buffer solution and with the force perturbation. The error bar in both figure 3.6C and 3.6F are forth central moment of FRET distribution. Evidently, under the force perturbation, the enzyme conformation is less sensitive to enzyme-substrate interactions comparing to the results in 3.6C when the measurement is under no external force perturbation. Our results demonstrate that the enzymatic conformational fluctuation accessible space is strongly influenced by the enzyme-substrate interactions, which provides experimental evidence showing the critical role of the protein-ligand interactions in a possible conformation selection mechanism of enzyme-substrate complex formation. Conformations of protein molecules undergo dynamical fluctuations under physiological conditions, while the existence of ligands induces conformational regulation energetically and spontaneously favor to those conformations involving in ligand-active sites binding interactions.76-79 Consequentially, the conformational distribution narrows down to a ligand-binding accessible conformational subset out of the broad conformational distribution of the apo HPPK enzymes without involving in protein-ligand interactions, as shown in Figure 3.6C and 3.6F. 75 Figure 3.7. 3 Conform mational flu uctuation raate distributtions calcullated from aautocorrelattion analysis of HPPK with w substraate ATP and d HP added d. (A) Autoccorrelation ffunctions frrom ntensity trajectory meaasured underr the condittion of with hout magnettic pulling FRET in force. Green line in ndicates don nor while reed for accep ptor. (B) Au utocorrelatio on function ns RET intensitty trajectory y measured under the ccondition off with magn netic pulling from FR force app plied. Green line indiccates donor while red fo for acceptorr. (C and D) Fluctuatio on rate distrributions off FRET don nor under co onditions w with and with hout magneetic pulling force. (E E and F) Fluctuation raate distributtions of FR RET accepto or under con nditions witth and with hout magnettic pulling force. f 76 3.3.4. Conformational Dynamics Manipulation by Single Molecule FRET-Magnetic Tweezers Spectroscopy To further characterize the changes in conformational dynamics with respect to the conformational flexibility and the accessibility of the conformations associated with the enzyme-substrate interactions, we analyze the autocorrelation functions of fluorescence fluctuation trajectories of our single molecule FRET measurements (Figure 3.7A and 3.7B), under the conditions of with and without the magnetic field, for both donor (Figure 3.7C and 3.7D) and acceptor (Figure 3.7E and 3.7F), while the HPPK molecule is with the substrate of ATP and HP added in the PBS buffer solution. Conformational fluctuation rate can be calculated from the exponential decay rate of autocorrelation function, and the essentially same decay rates between the autocorrelation functions of donor and acceptor in both A and B strongly indicate that the fluctuations are from the same origin, the single-molecular FRET. We have studied 30 different timing on FRET trajectories of one single molecule under both with and without magnetic pulling force conditions to have the distributions of conformational fluctuation rate as shown in Figure 3.7C, 3.7D, 3.7E and 3.7F. Figure 3.7C and 3.7D show the distributions of conformational fluctuation rate calculated from autocorrelation functions, and the distributions show a remarkable broadening, under the condition of with the magnetic pulling force, comparing to that of measured under without the magnetic pulling force. This result indicates an increase in distribution range of conformational fluctuation rate under the magnetic force pulling. Similar change triggered by magnetic tweezers pulling force also occurs consistently in acceptor fluctuation rate distributions (Figure 3.7E and 3.7F). 77 Such broadenings in the conformational fluctuation rate distribution are consistent with the results from the standard deviation analysis of FRET efficiency distributions. According to Figure 3.6C and 3.6F, it is the substrate binding interaction that leads to less flexible conformational fluctuations of HPPK protein molecule, while such interaction is weakened by applied external pulling force. Consequently, weakened enzyme-substrate interactions also result in an apparent broadening of conformational fluctuation rate distribution. Enzyme-substrate interaction is highly sensitive to the protein conformational perturbation by external pulling force. With the force pulling, such enzyme-substrate interaction is perturbed and weakened, releasing the protein from being constrained by ligand-binding interaction, resulting in a broader range of the enzyme conformational fluctuation rate. Our results show that the small external force of about 1.4±0.4 pN can apparently impact the enzymatic conformational fluctuation distribution, and the enzymatic conformational flexibility or conformational fluctuation distribution is critical for enzyme-substrate interaction, thus the small external force can impact the interactions between enzyme and substrate. This low external pulling force is not sufficient to rupture the protein tertiary structures as the rupture force is at least 18 pN for HPPK55 or even not sufficient to break hydrogen bonds as a typical hydrogen bonding force is about 4 pN and higher.80 Therefore, the low external force we applied to an individual HPPK enzyme molecule likely only causes a deformation of tertiary structure of the HPPK enzyme molecule. 78 To illustrate the fact that the small external pulling force is capable of impacting the enzymatic function, such as enzyme-substrate interaction, we note that the external pulling force applied on an single enzyme molecule through the magnetic tweezers, even if the force is at similar scale competing with the thermal fluctuation forces, is an onedirection constant force that capable of deviating the conformational fluctuation energy landscape, leading to a deformation of the HPPK enzyme molecule. As an analogy, it is simply like a random walk on a tilted energy landscape by an external and constant force field. Evidently, the one-direction pulling force decreases enzymatic conformational flexibility and affecting the enzyme-substrate interaction impacting enzymatic function. Furthermore, we emphasize that this work only focuses on the understanding of the enzyme-substrate interactions in forming the enzyme-substrate reactive complex, which is the first step of an enzymatic reaction, and our future work will focus on identify and characterizing the impact of the external force manipulation on the enzymatic reaction turnover activities. 3.4. Conclusion In summary, we have demonstrated that our correlated single-molecule FRETmagnetic tweezers microscopy is capable of manipulating the conformation of single enzyme molecules, and in turn, manipulating the enzyme-substrate interactions, by applying and controlling a pulling force on single kinase molecules. Technically, the correlated magnetic tweezers single-molecule FRET spectroscopy is a potentially powerful tool to interrogate the protein conformational dynamics and the associated protein functions. Using our approach, we are able to interrogate the conformational selection mechanism by exam the conformation flexibility and conformational fluctuation 79 accessible space when the enzyme is under interacting and not interacting with the substrate molecules. We have observed that the enzyme-substrate interaction provides a strong conformational selection effect through a folding-binding interacting process shifting the conformational fluctuation to more confined spatial range; whereas, under the force pulling, distorted enzyme conformation has a weaker interaction with the substrate, leading to a weak conformational selection effect and folding-binding interacting dynamics. 3.5 References 1 Schuler, B.; Eaton, W.A. Protein Folding Studied by Single-Molecule FRET. Curr. Opin. in Struct. Biol. 2008, 18, 16-26. 2 Lu, H.P. Science Enzymes in Coherent Motion. 2012, 335, 300-301. 3 Lu, H.P.; Iakoucheva, L.M.; Ackerman, E.J. Single-Molecule Conformational Dynamics of Fluctuating Noncovalent DNA-Protein Interactions in DNA Damage Recognition. J. Am. Chem. Soc. 2001, 123, 9184-9185. 4 Guha, S.; Sahu, K.; Roy, D.; Mondal, S. K.; Roy, S.; Bhattacharyya, K. Slow Solvation Dynamics at the Active Site of an Enzyme: Implications for Catalysis. Biochemistry 2005, 44, 8940-8947. 5 Zhang, Q.; Stelzer, A.C.; Fisher, C.K.; Al-Hashimi, H.M. Visualizing Spatially Correlated Dynamics that Directs RNA Conformational Transitions. Nature 2007, 450, 1263 – 1267. 6 Mittermaier, A.K.; Kay, L.E. Observing Biological Dynamics at Atomic Resolution Using NMR. Trends Biochem. Sci. 2009, 34, 601 – 611. 80 7 Pan, R.; Zhang, X. J.; Zhang, Z. J.; Zhou, Y.; Tian, W. X.; He, R. Q. SubstrateInduced Changes in Protease Active Site Conformation Impact on Subsequent Reactions with Substrates. J. Biol. Chem. 2010, 285, 22948 – 22954. 8 Wang, J.; Oliveira, R. J.; Chu, X.K.; Whitford, P.C.; Chahine, J.; Wei, H.; Wang, E.K.; Onuchic, J. N.; Leite, V. B.P. Topography of Funneled Landscapes Determines the Thermodynamics and Kinetics of Protein Folding. Proc. Natl. Acad. Sci. USA 2012, 109, 15763–8. 9 Puchner, E.M.; Gaub, H.E. Single-Molecule Mechanoenzymatics. Annu. Rev. Biophys. 2012, 41, 497-518. 10 Gumpp, H.; Puchner, E. M.; Zimmermann, J. L.; Gerland, U.; Gaub, H. E.; Blank, K. Triggering Enzymatic Activity with Force. Nano Lett. 2009, 9, 3290–3295. 11 Kuoa, T.; Garcia-Manyesb, S.; Lic, J.; Bareld, I.; Lue, H.; Bernec, B. J.; Urbakhd, M.; Klafter, J.; Fernández, J.M. Probing Static Disorder in Arrhenius Kinetics by Single-Molecule Force Spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 11336–11340. 12 Metzler, R.; Klafter, J. The Random Walk's Guide to Anomalous Diffusion Phys. Rep. 2000, 339, 1–77. 13 Mo, Y.; Bao. P.; Gao, J. Energy Decomposition Analysis Based on a BlockLocalized Wavefunction and Multistate Density Functional Theory. Phys. Chem. Chem. Phys. 2011, 13, 6760-6775. 14 Stirnemanna, G.; Kang, S.; Zhou, R.; Berne, B. J. How Force Unfolding Differs From Chemical Denaturation. Proc. Natl. Acad. Sci. USA 2014, 111, 3413–3418. 81 15 Zhou, R.; Berne, B. J.; Germain, R. The Free Energy Landscape for β Hairpin Folding in Explicit Water. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 14931-14936. 16 Kishino, A.; Yanagida, T. "Force measurements by micromanipulation of a single actin filament by glass needles". Nature 1988, 334 (6177): 74–76. 17 Sambongi, Y.; Iko, Y.; Tanabe, M.; Omote, H.; Iwamoto-Kihara, A.; Ueda, I.; Yanagida, T.; Wada, Y.; Futai, M. Mechanical Rotation of the c Subunit Oligomer in ATP Synthase (F0F1): Direct Observation. Science 1999, 286,1722-1724. 18 Cao, J. Chem. Event-Averaged Measurements of Single-Molecule Kinetics. Phys. Lett. 2000, 327, 38–44. 19 Yang S.; Cao, J. Direct Measurements of Memory Effects in Single-Molecule Kinetics. J. Chem. Phys. 2002, 117, 10996-11009. 20 Hinterdorfer, P.; Baumgartner, W.; Gruber, H. J.; Schilcher, K.; Schindler, H. Detection and Localization of Individual Antibody-Antigen Recognition Events by Atomic Force Microscopy. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 3477–3481. 21 Hinterdorfer, P.; Dufrêne, Y.F. Detection and Localization of Single Molecular Recognition Events Using Atomic Force Microscopy. Nat. Methods. 2006, 3, 347 – 355. 22 Raible, M.; Evstigneev, M.; Bartels, F. W.; Eckel, R.; Nguyen-Duong, M.; Merkel, R.; Ros, R.; Anselmetti, D.; Reimann, P. Theoretical Analysis of Single-Molecule Force Spectroscopy Experiments: Heterogeneity of Chemical Bonds. Biophys. J. 2006, 90, 3851-3864. 23 Schwesinger, F.; Ros, R.; Strunz, T.; Anselmetti, D.; Güntherodt, H.; Honegger, A.; Jermutus, L.; Tiefenauer, L.; Plückthun, A. Unbinding Forces of Single Antibody 82 Antigen Complexes Correlate with Their Thermal Dissociation Rates. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 9972-9977. 24 Fazal, F. M.; Block, S.M. Optical Tweezers Study Life Under Tension. Nat. Photon. 2011, 5, 318–321. 25 Svoboda, K.; Mitra, P.P.; Block, S.M. Fluctuation Analysis of Motor Protein Movement and Single Enzyme Kinetics. Proc. Natl. Acad. Sci. U.S.A. 1994, 91, 11782–11786. 26 Allen, S.; Chen, X.; Davies, J.; Davies, M.C.; Dawkes, A.C.; Edwards, J.C.; Roberts, C.J.; Sefton, J.; Tendler, S.J.B.; Williams, P.M. Detection of Antigen−Antibody Binding Events with the Atomic Force Microscope. Biochemistry, 1997, 36, 7457–7463. 27 Fotiadisa, D.; Scheuringa, S.; Müllera, S.A.; Engela, A.; Müller, D.J. Imaging and Manipulation of Biological Structures with the AFM. Micron 2002, 33, 385–397. 28 Choi, Y.; Moody, I.S.; Sims, P.C.; Hunt, S.R.; Corso, B.L.; Seitz, D.E.; Blaszcazk, L.C.; Collins, P.G.; Weiss, G.A. Single Molecule Dynamics of Lysozyme Processing Distinguishes Linear and Cross-linked Peptidoglycan Substrates. J. Am. Chem. Soc. 2012, 134, 2032-2035. 29 Anand, U.; Mukherjee, S. Reversibility in Protein Folding: Effect of β-cyclodextrin on Bovine Serum Albumin Unfolded by Sodium Dodecyl Sulphate. Phys. Chem. Chem. Phys. 2013, 15, 9375-9383. 30 Mojumdar, S. S.; Chowdhury, R.; Chattoraj, S.; Bhattacharyya, K. Role of Ionic Liquid on the Conformational Dynamics in the Native, Molten Globule, and 83 Unfolded States of Cytochrome C: A Fluorescence Correlation Spectroscopy Study. J. Phys. Chem. B, 2012, 116, 12189–12198. 31 Marszalek, P. E.; Lu, H.; Li, H.; Carrion-Vazquez, M.; Oberhauser, A. F.; Schulten, K.; Fernandez, J.M. Mechanical Unfolding Intermediates in Titin Modules. Nature 1999, 402, 100-103. 32 Bustamante, C.; Chemla, Y.R.; Moffitt, J.R. Single Molecule Techniques Cold Spring Harbor Laboratory Press: New York, 2008; 297-325. 33 Lipman, E. A.; Schuler, B.; Bakajin, O.; Eaton, W.A. Single-Molecule Measurement of Protein Folding Kinetics. Science 2003, 301, 1233-1235. 34 Neuman, K. C.; Nagy, A. Single-Molecule Force Spectroscopy: Optical Tweezers, Magnetic Tweezers and Atomic Force Microscopy. Nat.Methods 2008, 5, 491-505. 35 Rief, M.; Oesterhelt, F.; Heymann, B.; Gaub, H.E. Single Molecule Force Spectroscopy on Polysaccharides by Atomic Force Microscopy. Science. 1997 275(5304): 1295-1297. 36 Lee, S.; Hohng, S. An Optical Trap Combined with Three-Color FRET. J. Am. Chem. Soc. 2013, 135, 18260–18263 37 Haber, C.; Wirtz, D. Magnetic Tweezers for DNA Micromanipulation. Rev. Sci Instrum. 2000, 71, 4561-4570. 38 Tanase, M.; Biais, N.; Sheetz, M. In Cell Mechanics, edited by Wang, Y. L. and Discher, D. E. 2007; 83, 473-493. 39 Fisher, J. K.; Cummings, J.R.; Desai, K. V.; Vicci, L.; Wilde, B.; Keller, K.; Weigle, C.; Bishop, G.; Taylor, R. M.; Davis, C. W.; Boucher, R. C.; O'Brien, E. T.; Superfine, R. Three-Dimensional Force Microscope: A Nanometric Optical Tracking 84 and Magnetic Manipulation System for the Biomedical Sciences. Rev. Sci Instrum. 2005, 76, 053711. 40 Kollmannsberger, P.; Fabry, B. High-Force Magnetic Tweezers with Force Feedback for Biological Applications. Rev. Sci Instrum. 2007, 78, 114301. 41 Kruithof, M.; Chien, F.; de Jager, M.; van Noort, J. Subpiconewton Dynamic Force Spectroscopy Using Magnetic Tweezers. Biophys. J. 2008, 94, 2343-2348. 42 Leuba, S.H.; Karymov, M.A.; Tomschik, M.; Ramjit, R.; Smith, P.; Zlatanova, J. Assembly of Single Chromatin Fibers Depends on the Tension in the DNA Molecule: Magnetic Tweezers Study. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 495500. 43 Ribeck, N.; Saleh, O. A. Multiplexed Single-Molecule Measurements with Magnetic Tweezers. Rev. Sci Instrum. 2008, 79, 094301. 44 Smith, S. B.; Finzi, L.; Bustamante, C. Direct Mechanical Measurements of the Elasticity of Single DNA Molecules by Using Magnetic Beads. Science 1992, 258, 1122-1126. 45 Yan, J.; Skoko, D.; Marko, J.F. Near Field Magnetic Tweezer Manipulation of Single DNA Molecules. Phys. Rev. E 2004, 70, 011905. 46 Gosse, C.; Croquette, V. Magnetic Tweezers: Micromanipulation and Force Measurement at the Molecular Level. Biophys. J. 2002, 82, 3314-3329. 47 Strick, T.R.; Allemand, J.F.; Bensimon, D.; Bensimon, A.; Croquette, V. The Elasticity of A Single Supercoiled DNA Molecule. Science. 1996 271(5257): 18351837. 85 48 del Rio, A.; Perez-Jimenez, R.; Liu, R.; Roca-Cusachs, P.; Fernandez, J.M.; Sheetz, M.P. Stretching Single Talin Rod Molecules Activates Vinculin Binding. Science. 2009 323 (5914): 638-641. 49 Liu, R.C.; Garcia-Manyes, S.; Sarkar, A.; Badilla, C.L; Fernandez, J.M. Mechanical Characterization of Protein L in the Low-Force Regime by Electromagnetic Tweezers/Evanescent Nanometry. Biophys. J. 2009, 96, 3810-3821. 50 Blaszczyk, J.; Shi, G.; Yan, H.; Ji, X. Catalytic Center Assembly of HPPK as Revealed by the Crystal Structure of a Ternary Complex at 1.25 Å Resolution. Structure 2000, 10, 1049-1058. 51 Stammers, D. K.; Achari, A.; Somers, D. O.; Bryant, P. K.; Rosemond, J.; Scott, D. L.; Champness, J.N. 2.0 Å X-ray Structure of the Ternary Complex of 7,8-dihydro-6hydroxymethylpterinpyrophosphokinase from Escherichia coli with ATP and a Substrate Analogue. FEBS Lett. 1999, 456, 49. 52 Xiao, B.; Shi, G.; Chen, X.; Yan, H.; Ji, X. Crystal Structure of 6-hydroxymethyl7,8-dihydropterin Pyrophosphokinase, a Potential Target for the Development of Novel Antimicrobial Agents. Structure 1999, 5,489-496. 53 laszczyk, J.; Li, Y.; Wu,Y.; Shi, G..; Ji, X.; Yan, H. Essential Roles of a Dynamic Loop in the Catalysis of 6-hydroxymethyl-7,8-dihydropterin Pyrophosphokinase. Biochemistry 2004, 43, 1469. 54 He, Y.; Li, Y.; Mukherjee, S.; Wu, Y.; Yan, H.; Lu, H. P. Probing Single-Molecule Enzyme Active-Site Conformational State Intermittent Coherence. J. Am. Chem. Soc. 2011, 133, 14389-14395. 55 Note: the value is according to the product specification from Invitrogen Company. 86 56 He, Y.; Lu, M.; Cao, J.; Lu, H.P. Manipulating Protein Conformations by SingleMolecule AFM-FRET Nanoscopy. ACS Nano, 2011, 6, 1221-1229. 57 de Vries, A. H. B.; Krenn, B.E.; van Driel, R.; Kangersd, J.S. Micro Magnetic Tweezers for Nanomanipulation Inside Live Cells. Biophys. J. 2005, 88, 2137–2144. 58 Forth, D.; Sheinin, M.Y.; Inman, J.; Wang, M.D. Torque Measurement at the SingleMolecule Level. Annu. Rev. Biophys. 2013, 42, 583-604. 59 Hammes, G.G. Multiple Conformational Changes in Enzyme Catalysis. Biochemistry 2002, 41, 8221-8228. 60 Happel, J.; Sellers, P.H. New Perspective on the Kinetics of Enzyme Catalysis. J. Phys. Chem. 1995, 99, 6595-6600. 61 Hammes-Schiffer, S.; Benkovic, S.J. Relating Protein Motion to Catalysis. Annu. Rev. Biophys. 2006, 75, 519-541. 62 Min, W.; Xie, X. S.; Bagchi, B. J. Role of conformational dynamics in kinetics of an enzymatic cycle in a nonequilibium steady state. Chem. Phys. 2009, 131, 065104: 16. 63 Benkovic, S.J.; Hammes-Schiffer, S. A Perspective on Enzyme Catalysis. Science 2003, 301, 1196-1202. 64 Garcia-Viloca, M.; Gao, J.; Karplus, M.; Truhlar, D.G. How Enzymes Work: Analysis by Modern Rate Theory and Computer Simulations. Science 2004, 303, 186-195. 65 Chu, X.; Gan, L.;Wang, E.; Wang, J. Quantifying the Topography of the Intrinsic Energy Landscape of Flexible Biomolecular Recognition. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, E2342-51. 87 66 Shoemaker, B.A.; Portman, J.J.; Wolynes, P.G. Speeding Molecular Recognition by Using the Folding Funnel: The Fly-Casting Mechanism. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 8868-8873. 67 Antikainen, N. M.; Smiley, R. D.; Benkovic, S. J.; Hammes, G.G. Conformation Coupled Enzyme Catalysis: Single-Molecule and Transient Kinetics Investigation of Dihydrofolate Reductase. Biochemistry 2005, 44, 16835–16843. 68 Eisenmesser, E. Z.; Millet, O.; Labeikovsky, W.; Korzhnev, D. M.; Wolf-Watz, M.; Bosco, D. A.; Skalicky, J. J.; Kay, L. E.; Kern, D. Intrinsic dynamics of an enzyme underlies catalysis. Nature 2005, 438, 117–121. 69 Henzler-Wildman, K. A.; Thai, V.; Lei, M.; Ott, M.; Wolf-Watz, M.; Fenn, T.; Ed Pozharski, Wilson, M. A.; Petsko, G. A.; Karplus, M.; Hübner, C. G.; Kern, D. Intrinsic Motions Along an Enzymatic Reaction Trajectory. Nature 2007, 450, 838844. 70 Pisliakov, A.V.; Cao, J.; Kamerlin, S. C. L.; Warshel, A. Enzyme millisecond conformational dynamics do not catalyze the chemical step. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 17359–17364. 71 Whitford, P. C.; Onuchic J. N.; Wolynes, P. G. Energy landscape along an enzymatic reaction trajectory: hinges or cracks? HFSP J. 2008, 2, 61–64. 72 Xie, X.S. Enzyme Kinetics, Past and Present. Science 2013, 342(6165), 1457-1459. 73 Watt, E.D.; Shimada, H.; Kovrigin, E. L.; Loria, J.P. The Mechanism of RateLimiting Motions in Enzyme Function. Proc. Natl. Acad. Sci. U.S.A. 2007,104, 11981–11986. 88 74 Kalinin, S.; Sisamakis, E.; Magennis, S.W.; Felekyan, S.; Seidel, C.A.M. On the Origin of Broadening of Single-Molecule FRET Efficiency Distributions beyond Shot Noise Limits. J. Phys. Chem. B 2010, 114, 6197–6206. 75 We note that there are multiple origins such as triplet excitation, blinking, or heterogeneity of fluorophore may contribute to the standard deviation of a FRET efficiency distribution. However, our experimental measurements are performed under the same optical conditions, and the differences are only the non-optical parameters, such as force pulling or no force pulling, and with substrate and without substrate in solutions. And the changes of FRET standard deviation in our results are reproducible over time as shown in Figure 3.6C and 3.6F. 76 Long, D.; Bruschweiler, R.J. Atomistic Kinetic Model for Population Shift and Allostery in Biomolecules. J. Am. Chem. Soc. 2011, 133, 18999-19005. 77 Goh, C.; Milburn, D.; Gerstein, M. Conformational Changes Associated with Protein–Protein Interactions. Curr. Opin. in Struct. Biol. 2004, 14, 1-6. 78 James, L. C.; Tawfik, D.S. Conformational Diversity and Protein Evolution – a 60Year-Old Hypothesis Revisited. Trends Biochem Sci. 2003, 28, 361-368. 79 Okazaki, K. I.; Takada, S. Dynamic Energy Landscape View of Coupled Binding and Protein Conformational Change: Induced-Fit versus Population-Shift Mechanisms. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 11182-11187. 80 Finer, J.T.; Simmons, R.M.; Spudich, J.A. Single Myosin Molecule Mechanics: Piconewton Forces and Nanometre Steps. Nature 1994, 368, 113 - 119. 89 CHAPTER IV. INTERROGATING THE ACTIVITIES OF CONFORMATIONAL DEFORMED ENZYME B BY SINGLE MOLECULE TIRF-MAGNETIC TWEEZERS MICROSCOPY 4.1 Introduction One of the central focuses in protein study is the structure-function relationship, the impact of different conformations to the properties of protein molecules. There has been intensive research reported on that protein molecules with their tertiary structure perturbed or even partially unfolded may be related to misfunction or causing diseases, because changing protein conformations typically leads to significant differences in their affinity, selectivity, and reactivity. 1-22 In modern enzymology, it has been extensively explored that the enzymatic conformation-function relationship, especially in the dynamic rather than the static perspectives, plays a critical role to understand enzyme mechanism at molecular level. 23-28 For example, in an enzymatic reaction, forming enzyme–substrate reactive complex often involves significantly enzymatic active site conformational changes, being a critical step in defining enzymatic reaction potential surface, reaction transition state, and reaction pathways. Such enzyme–substrate interaction process has been demonstrated to be significantly affected by enzyme conformation deformations. 1,4 18-20 Traditional enzyme studies focused on enzyme molecules at enzymatic reaction conditions while the enzymes are fully folded or in their natural states. Such research works on enzymatic stability studies focused on ensemble level activity of enzyme at different physical conditions or chemical environment without probing corresponding change in conformation of enzyme molecules.29-37 In recent years, more and more researches have focused on studying 90 enzymes and their activities at their deformed, unfolded, or force-manipulated states.3,7,38-40 Typically, such experiments on enzymes are under non-physiological or non-enzymatic reaction conditions, or even under denatured conditions under which the enzyme unfolds.41-44 On the other hand, conformational manipulation on single protein molecule by applying mechanical force has been achieved by several different approaches. For example, Atomic Force Microscopy (AFM) have been widely used to study conformational dynamics of single protein molecules;45-48 Optical tweezers has been applied to study folding pathway or even conformational folding transition state of single protein molecules;49-51 Magnetic tweezers has also been explored to study protein conformational dynamics.40,52 Recently, rupturing a single enzyme molecule and observing its recovery of activity under enzymatic reaction conditions has been achieved using AFM.7 However, manipulating conformation of a single enzyme molecule in its deformed and partially unfolded states under a physiological enzymatic reaction condition with simultaneous observation of its activity remains a challenge, exploring how critical the enzyme conformational stability as well as dynamically fluctuating and externally force perturbed enzymatic states impact on the enzymatic activities. A single molecule level observation of enzymatic reactivity in the contest of conformational deformation of the enzyme protein molecules will provide us a fundamental understanding of the dependence of enzymatic reactivity on the conformational changes and stability. The impact of different conformation on enzyme function has also been the focus of theoretical studies.23-24, 5355 A range of key questions on how the enzymes work can be investigated. For example, does a conformation-deformed or even partially unfolded enzyme molecule still have measurable enzymatic reactivity? If so, how much activity will be left at various degree of external force perturbation? And in terms of molecular folded conformations, how much can an enzyme 91 molecule tolerate such conformational deformation under an enzymatic reaction condition, i.e., under enzyme-substrate binding interaction conditions? Here we report our work towards obtaining the answers for these questions. In Chapter III, we have investigated that enzyme–substrate interaction induced enzymatic active site fluctuation dynamics and flexibility associated with induced fit and folding-binding mechanism by manipulating the enzyme conformations and fluctuations at single molecule level using our home developed single molecule FRET magnetic tweezers which generating forces at 1-10 picoNewton scale.52 Here we report our new approach to manipulate single molecule catalytic activity using magnetic tweezers to deform the conformation of horseradish peroxidase (HRP) enzyme at single molecule level, correlated with total internal reflection (TIRF) microscope to simultaneously observe the fluorogenic enzymatic turnovers, as shown in Figure 4.1. There are specific advantages of using magnetic tweezers to provide an external mechanical force to manipulate single molecule enzyme, including, (1) wide force range from less than hydrogen bonding force to protein rupture force; (2) no photo-damage and cross talk to single molecule spectroscopic measurements of enzymatic activity and enzyme conformational changes; and (3) capability to simultaneously applying force on a large number of single molecules.38-40,56-59 Combined with TIRF microscopy as optical measurement, it provides us the unique capability and opportunity to interrogate conformation-function relationship of enzyme molecules under enzymatic reaction conditions, specifically studying the impact of deforming protein conformation on protein function at single molecule level. 92 4.2 Mateerials and Methods M 4.2.1. Ma aterials. Horseradish H peroxidase p (H HRP) is a 34 4 kDa 306-reesidue monoomeric enzym me. The HR RPcatalyzed d reaction co onverts hydro ogen peroxid de (H2O2) annd non-fluorrescent probee substrate N Nacetyl-3,7 7-dihydroxy yphenoxazinee (APR) into o fluorescentt resorufin. This enzym matic reactionn is fluorogen nic, in which h only the reeleased produ uct moleculees emit fluorrescence thatt is detectablle by total internal reflectio on fluorescence microscopy. In our experiment,, we choose supernetic beads (Dynabeads® ( ® MyOne™ ™ Streptavidiin T1 Invitroogen Companny) with 1.005 paramagn µm diam meter, covalen ntly attachin ng the beads to biotin co--factor linkinng to the HR RP molecules through a biotin-strep ptavidin link k. The HRP enzyme moolecules are iin substrate ssolution consistin ng of 50 mM PBS buffer solution (pH H =7.4), 1000 nM N-acetyyl-3,7dihydrox xyphenoxazin ne (Amplex Red, APR) and 100 nM M hydrogen pperoxide (H2O2). 93 Figure 4.1. A conceptual scheme of our experimental system. Horseradish peroxidase (HRP) molecules are tethered at one end to a modified glass coverslip. The immobilized protein molecules are tethered at the other end to magnetic beads through biotin-streptavidin linking. 4.2.2. TIRF Measurement. TIRF measurements are carried out by using an inverted confocal microscope (Olympus IX 71 with 60 x objective) with a 532 nm CW crystal laser generating evanescent wave for total internal excitation. Emitted signal is filtered with a long-pass beam splitter and collected by an Electron Multiplying Charge Coupled Device (EMCCD: ProEM 512B, PI co.). We conduct the single molecule total internal reflection optical measurements and pulling manipulation via magnetic tweezers simultaneously. Magnetic force was applied through those attached superparamagnetic beads on HRP molecules. The essential component of our magnetic tweezers device is a permanent magnet generating magnetic field, which was mounted on a specially made stage enabling the magnetic probe to move along any direction and for any desired distance. The sample chamber was put on an x-y stage capable of applying in-plane adjustment. The distance between the magnet and the sample cover glass is 4 mm, implying an 1100 Gauss magnetic field at the sample. In our experiments, we applied mechanical force to single enzyme molecules by applying external magnetic field and sensing through tethered magnetic bead on those single enzyme molecules. Quantitative calculation of the force generated by our magnetic tweezers has been discussed in Chapter 3.2.4. In brief, pulling force roughly at 1-3 pN can be applied on the targeted single HRP protein molecules using our magnetic tweezers setup. Although the force is 94 weaker than hydrogen bonding force which is typically 6-9 pN, we have demonstrated that force at this scale is capable to trigger conformational response in our previous publication.52 4.2.3. Sample Preparation. As shown in Figure 4.1, the HRP molecules were bound to the glass cover slip at one end by 3-aminopropyltriethoxy-silane (TESPA)-Dimethyl Suberimidate•2HCl (DMS) linkers and linked to a streptavidin coated superparamagnetic bead (Dynabeads® MyOne™ Streptavidin T1 Invitrogen Company), at the other end via biotin-streptavidin bond. Protein immobilization was carried out through a routine procedure as shown in Figure 4.2. In brief, a clean glass coverslip was firstly immersed overnight in NaOH-ethanol solution, and the coverslip was next washed by distilled water, blow-dried by air flow, and incubated with a DMSO solution containing a mixture in 10% concentration consisting of TESPA and isobutyltrimethoxysilane in 1:10000 ratio overnight. The coverslip was then washed by distilled water and consecutively transferred and incubated for 4 hours in each system below: 15 mL PBS buffer solution pH=8.0, containing 10nM Dimethyl Suberimidate•2HCl (DMS•2HCl); 15 mL PBS buffer solution pH=7.4, containing 10 nM HRP; 15 mL PBS buffer solution pH=7.4, containing 10 nM NHSPEO12-biotin; 15 ml PBS solution pH=7.4, containing 1µl magnetic beads stock solution which is commercial available. The low concentration of each solution was to make sure that the distribution of the individual enzyme molecules on cover glass is adequately separated so that one bead does not attach to multiple enzyme molecules. Meanwhile, low concentrations of TESPA are used to ensure that immobilized protein molecules are distributed separately enough from each other for obtaining single molecule TIRF images. 95 Figure 4.2. 4 Preparatiion of singlee molecule HRP H sample. We tethereed protein moolecules at oone end to the coverslip by b Dimethyll suberimidatte-2HCl (DM MS-2HCl) annd at the othher end to a 1 µm size paramagnetic beead by biotin n-streptavidin n bonding. F Force was im mplied by adding externaal magneticc field and heence the mollecule could d feel it throuugh the beadds. We W also havee noted that either e biotin or DMS cann only be teth thered to a H HRP moleculle via connectio on with lysin ne in the amiino acid sequ uence, whichh leads to m multiple possiible tetheredd condition n of the proteein moleculee to coverslip p or magnetiic beads. Thhere are six llysine residuues on one HRP H moleculle: residue 65 5, 84, 149, 174, 1 232, 2411. In our expperiment, eaach HRP moleculee are immobiilized on cov verslip via DMS-lysine D ccovalent linkk at one out of these six lysine ressidues, and being b tethereed to magnettic beads thrrough anotheer lysine resiidue out of thhe 96 rest five options. Therefore, in our sample preparation process, each HRP molecules can be immobilized by using any two of the total six lysine residues to link toward DMS and biotin. There are 15 possible lysine combinations in total, leading to 15 possible different tethering conditions. Since we are not able to pinpoint that a pair of specific lysine residues tethered to a specific protein molecule, we have performed Steered Molecular Dynamics (SMD) simulation for all 15 possible tethering conditions for HRP molecule, showing that when being stretched by external force, the active site of HRP molecule will be deformed beyond the scale of its normal fluctuation in most of possible conditions. Details of the SMD simulation will be discussed in chapter V. 4.3. Results 4.3.1. Single-Molecule TIRF Imaging Measurement of HRP Activity Figure 4.3B shows the time trajectories of fluorescence intensity of a single HRP molecule under a fluorogenic enzymatic assay condition. Time resolution of our TIRFM imaging measurement is 20ms per frame, while each measurement lasts 60 seconds and accumulates totally 3000 imaging frames with 10 ms data readout time for each frame. The fluorescence signals we take into account as enzymatic turnover events are the photon count spikes above the threshold of the trajectory. The threshold is set three times the standard deviation larger than the distribution mean value of the histogram deduced from the whole trajectory over time domain. In brief, we firstly fit the TIRF experimental real time trajectories by Gaussian distribution. Then we set the threshold value of signal from real time trajectory as ‘higher than three times the standard deviation above the mean value of overall trajectory’ part. 97 In this way, we have the confiden nce level at least l larger tthan 95% to discern signnals from backgrou und noise. Figure 4.3. 4 Single-tu urnover detecction of HRP P enzyme caatalysis. (A) Exemplary fluorescencee time trajeectory of sin ngle HRP mo olecule obserrved from TIRFM measurement. (B B) Timedistributiion of the TIIRFM trajecttory. On tim me domain, siignals abovee threshold aare taken intoo account as a turnover events. e (C) Segment of the TIRFM turnover trajjectory. τofff is the waitinng time betw ween sequen ntial reaction n events. 4.3.2. An nalysis of Sin ngle-Molecu ule Activity Trajectories T s Measured u under Forcee Pulling an nd Releasing g Condition ns. We W analyzed 30 individuaal moleculess under the eexternal forcce manipulation of pullinng and releaasing, shown n in Figure 4.4. The HRP P enzyme moolecules are in 50 mM P PBS buffer solution (pH ( =7.4) with w Amplex Red (100 nM M) and H2O2 (100 nM) aas substratess. We apply 1100 Gau uss magneticc field to gen nerate appro oximately 1.55pN pulling force for “pulling”, whiile not apply ying any field for “releassing”. Figurre 4.4A and 44.4B show thhat when beeing pulled bby external magnetic m forrce, the num mber of turno over events ooccurred on tthose HRP m molecules evidently y decrease, in ndicating a decrease d of catalytic c actiivity of thosee HRP enzym me moleculees. 98 Figure 4.4. 4 Histogram m results off turnover events from 300 individual HRP molecuules. (A) Turnoverr events histo ogram when n no force is applied on H HRP molecuules, the “releasing” grouup of HRP. (B)) Turnover events e histog gram when th he HRP mollecules are ppulled by appplied force frrom magneticc tweezers, th he “pulling”” group of HR RP. To T further qu uantitatively characterizee the impact of conformaational distorrtions on thee enzymatiic activity off HRP moleccules by usin ng the mechaanical force manipulatioon, we analyzze both the distribution of the turnov ver waiting time t and disstribution of detected phooton from released products bassed on the siingle-molecu ule fluorogennic trajectoriies recordedd, as shown iin Figure 4..5. The turnover waiting g time, τoff, is the time innterval betweeen two conssecutive deteected fluorogen nic turnover events, abov ve-threshold d fluorescencce signal inteensity jumpss. The turnoover waiting time, the tim me that needeed for actual events of caatalytic produucts formation, is negatiively 99 proportional to the enzymatic reactivity. In an enzymatic reaction, product formation rates are determined by the rate of substrate diffusion and enzyme-substrate complex formation, besides the reaction and releasing products. Although individual waiting time values are stochastic, the mean waiting time, <τoff>, and its distributions are defined by enzymatic reaction rate. In our experiment, we analyze the mean waiting time <τoff> over the turnover trajectories from each HRP enzyme molecule. When under pulling force via magnetic tweezers, deformation of the HRP enzyme occurs, the <τoff> accordingly increases due to the decrease of the time-averaged single-molecule catalytic rates of HRP enzyme, as shown in Figure 4.5A. We have further evaluated the enzymatic reaction activity changes under force pulling by counting photon bursts from the product releasing events. We calculate the integral area of fluorescence signal above the threshold by three times standard deviation higher than the mean value of the whole time trajectory. Figure 4.5B shows the distribution of the total photon counts from the product turnover events for each examined single molecule HRP under both force pulling and non-force pulling conditions. The total photon counts of enzymatic turnover photon burst events are calculated by counting all the photons above the threshold with and without magnetic pulling force for each single molecule HRP examined. Under the force pulling, the product burst counts decrease significantly in identical with the decreased number of turnover events, presumably associated with significant enzyme conformation deformation. This result of the enzymatic reaction activity decreases with the increase of the enzyme conformation deformation by force manipulation is consistent with the results of <τoff> analysis. To reveal that if the reduced activity is the result of change in substrate binding, we have further studied the change in enzyme-substrate binding affinity by calculating the equilibrium Dissociation Constant Kd, which is defined in equation 4.1 and equation 4.2. 100 koff ZZZX ES YZZ ZE+S k on Kd = koff kon = τ off τ on (4.1) (4.2) In equation 4.1, E and S stand for enzyme and substrate. Rate of binding and dissociation of enzyme-substrate complex are characterized by kon and koff respectively. Waiting time τoff and on-time τon are defined as shown in figure 4.5A. The result of Kd calculation is shown in Figure 4.5D, in which blue cubic spots stand for when the HRP enzyme molecules are released with no pulling force applied on them, and red triangle spots indicate the condition that ‘pulling force is applied by magnetic tweezers’ when the enzyme molecules are stretched and hence conformational deformed by external pulling force. We can find that when being stretched by external pulling force, the dissociation constant Kd become larger, indicating a weaker ligandsubstrate binding ability, which is as expected, since the enzyme conformation has been deformed. Although a deformed enzyme molecule can rely on fluctuation to come back to its active conformation, the stability will be affected to be less due to external force. 101 Figure 4.5. 4 Analysis of the relatiionship betw ween turnoveer event, meaan waiting tiime and prodduct burst of single s HRP molecules. m (A A) Correlatiion plots betw ween turnovver event couunts and meaan waiting time of each single HRP molecules with w and withhout magnettic pulling foorce appliedd. (B) Correlatiion plots betw ween turnov ver event cou unts and phooton burst coounts from reeleased prodducts of each single HRP molecules m wiith and witho out magneticc pulling forrce applied. (C) Correlattion plots betw ween turnov ver event cou unts and photon burst couunts from reeleased produucts of each single HR RP moleculees with and without w mag gnetic pullingg force appliied. (D) Enzzyme-substraate dissociation constantt of each sing gle HRP molecules withh and withouut magnetic ppulling forcee applied. 102 4.3.3. Repetitive Force Pulling-Releasing Manipulation of Enzyme Conformation for Impacting Enzymatic Activity. To further demonstrate the reproducibility and effectiveness of the force manipulation via magnetic tweezers correlated single-molecule TIRFM spectroscopy to the protein function, we measure the response of HRP enzymatic activity to repetitive force manipulation, toggling between “pulling” and “releasing” force applications, as shown in Figure 4.6. Under the “pulling” condition, 1100 Gauss magnetic field is applied to generate 1-3 pN mechanical force to deform the enzyme conformation; while under the “release” condition, there are no force is applied at all. In the experiment, the reaction system is first observed by TIRFM without any pulling force from the magnetic field for 100 seconds, and then, the pulling force is applied for the next 100 seconds; and the process is repeated for a few times for the next 300 seconds. Figure 4.6C shows that the total photon counts from the product turnover events of a single HRP molecule toggles between two different levels: reflecting the single molecule enzymatic activity changes due to the conformational manipulation by the external force pulling and releasing. Such response demonstrates the reproducible impact of the external force to the single enzyme catalytic function by affecting substrate binding process via deforming conformation. 103 Figure 4.6. 4 Responsee of HRP en nzymatic actiivity to repettitive magneetic pulling fforce. (A) (B B) Conceptu ual scheme of o HRP moleecule at releaased and pullled state, respectively. ((C) Product counts frrom 20 differrent HRP mo olecules with h and withouut being pullled. Errors aare estimatedd as 10% considering the possible inaaccuracies in nvolved in thhe setting thrreshold methhod in our daata analysis. 104 4.4. Discussion In our previous work, we have specifically analyzed an enzyme conformational change under the same experimental configuration and same magnetic field strength, and we have reported that the 1-3 pN external force can result that an enzyme from unfolding or deformation by 30% to even 100%, partially unfolded protein, while by applying such external pulling force, the enzyme-substrate binding interaction can be weakened, yet not completely diminished.52 We note that although the applied stretching force on enzyme protein molecules are weaker than hydrogen bonding force which is typically 6-9 pN, it is a one-direction constant force that is capable of deviating the conformational fluctuation energy landscape to deform conformation of single protein molecules. From the result in Figure 4.5 and Figure 4.6, we can see that when such external pulling force is applied, the product burst showing a significant decrease indicating the decreasing catalytic activity of HRP enzyme molecules. This change is as expected, since the conformation of HRP protein molecules are deformed or partially unfolded by external pulling force. We will also see from the steered MD simulation in Chapter 5 which also shows supporting result, illustrating the distortion of active site of a single HRP molecule when being pulled. In literature, there are studies on enzyme folding and unfolding under the overall denaturing solutions, where the enzyme either at unfolded condition but not enzymatic conditions or at the enzymatic reaction conditions but the enzyme is fully folded.60-69 In our experiment, we achieved actively manipulated single enzyme molecules to partially unfolded conformation under a physiological enzymatic reaction condition. To the best of our knowledge, our results present, for the first time, successfully manipulate protein function at single molecule level by 105 conformational manipulation that applying and controlling pulling force on single HRP molecules under physiological enzymatic reaction conditions with the existence of substrates. Revealing Significant Tolerance of Enzymatic Activity to Protein Conformational Deformations. It is also interesting that the HRP enzyme molecules are not totally lost their catalytic activity when their conformations are stretched by an external force, although less active than when in unperturbed condition, as shown in Figure 4.5 and Figure 4.6. In chapter 5, we will also see that MD simulation results also show a noticeable distortion of the active site in HRP molecule when the molecule is pulled by magnetic tweezers, yet chemical process in protein functioning such as electron transfer or proton transfer requires precision at Å in conformation. However, our result does not contradict the traditional structure-function relationship, because of conformational fluctuation of protein molecules. The critical factor is conformational fluctuation. When being deformed by external constant pulling force in our experiment, enzyme molecules are still capable to temporarily come back by conformational fluctuation to its substrate-binding accessible conformational subsets, although with a lower possibility compared with enzymes without pulling force applied on them. For enzyme molecules in enzymatic reaction conditions, such come-back from conformational fluctuation is most likely further facilitated by enzyme-substrate interactions. In enzymatic reactions, enzyme-substrate interaction plays a key role by affecting enzymatic active site conformation and induces the active site conformational fluctuation towards folded into active-favored subsets from foldingunfolding conformational fluctuation of enzyme molecules. The enzyme–substrate complex formation can regulate both static and dynamic conformations of the enzyme. In other words,k substrate-binding process induces the partially unfolded or deformed enzyme active site 106 conformation to be refolded to the active and nature conformation to produce the enzymatic reaction turnovers. A conceptual picture of this explanation is shown in Figure 4.7. Such understanding of constant conformational fluctuations of protein molecules are also in accordance with recent research of intrinsically disordered protein, or intrinsically unstructured protein. An intrinsically disordered protein typically does not fold in a well-defined three dimensional structure under physiological conditions.70-73 Such phenomena spurred extensive studies in recent years.74-84 Significantly different conformations can be detected for the same type of protein molecules in solution.85-87 Although being observed as lacking stable tertiary and/or secondary structure, intrinsically disordered proteins are capable to carry out specific functions. Our results here provides a possible mechanism for the existence of intrinsically disordered protein by revealing conformational fluctuation of enzyme protein molecules, and identifying that such conformational fluctuation can be regulated by ligand binding process. By substrate induced enzyme-substrate interaction, deformed enzyme molecules can fluctuate from ‘unstructured’ back to ‘structured’ state, still capable to maintain their function. Similarly, it is possible for intrinsically disordered protein molecules to fluctuate among different conformations to keep or quit from certain functions. 107 Figure 4.7. 4 Conceptu ual scheme of o conformattional fluctuuation of singgle enzyme pprotein whenn being defformed by ex xternal forcee. The circleed part conceeptually reprresents the aactive site onn a single en nzyme moleccule. When the t conformaation of an eenzyme moleecule is defoormed by extternal force, sub bstrate bindiing induced conformatio onal changess allow the ennzymatic active site to ccome back to itts active con nformation, leading l to occcurrence off reaction eveents. 4.5. Conclusion n this work, we have dem monstrated th hat using thee single-mollecule TIRF--magnetic In tweezers correlated im maging specctroscopy, th he conformattional perturrbations from m pulling forrce me function: Structural deformation d n of enzyme m molecules leead to can maniipulate enzym correspon nding chang ges in their acctivity. Such h influence rreveals that enzyme has a remarkablle countenaance and toleerance towarrd conformattional distorttion, i.e., thee enzyme cann still possesss significan nt activity ev ven the enzy yme conform mation in its ddeformed orr partially-unnfolded. Ouur experimeental approacch provides a unique cap pability: actiively manipuulating an ennzyme to 108 partially unfolded conformation under a physiological enzymatic reaction condition. Consequentially, we are able to interrogate the enzymatic reactivity in the contest of the protein structure-function relationship in a unique experiment. This repetitive manipulation of enzymatic activity reveals the capability of manipulate protein function by applying mechanical perturbation on its conformation. 4.6. References 1 Min, W.; Xie, X. S.; Bagchi, B. J. Role of Conformational Dynamics in Kinetics of an Enzymatic Cycle in a Nonequilibium Steady State. Chem. Phys. 2009, 131, 065104. 2 Chu, X.; Gan, L.; Wang, E.; Wang, J. Quantifying the Topography of the Intrinsic Energy Landscape of Flexible Biomolecular Recognition. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, E2342-2351. 3 Wang, J.; Oliveira, R. J.; Chu, X.K.; Whitford, P.C.; Chahine, J.; Wei, H.; Wang, E.K.; Onuchic, J. N.; Leite,V. B.P. Topography of Funneled Landscapes Determines the Thermodynamics and Kinetics of Protein Folding. Proc. Natl. Acad. Sci. USA 2012, 109, 15763.-15768 4 Whitford, P. C.; Onuchic, J. N.; Wolynes, P. G. Energy Landscape Along an Enzymatic Reaction Trajectory: Hinges or Cracks? HFSP J. 2008, 2, 61-64. 5 Shoemaker, B.A.; Portman, J.J.; Wolynes, P.G. Speeding Molecular Recognition by Using the Folding Funnel: The Fly-Casting Mechanism. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 8868-8873. 109 6 Puchner, E.M.; Gaub, H.E. Single-Molecule Mechanoenzymatics. Annu. Rev. Biophys. 2012, 41, 497-518. 7 Gumpp, H.; Puchner, E. M.; Zimmermann, J. L.; Gerland, U.; Gaub, H. E.; Blank, K. Triggering Enzymatic Activity with Force. Nano Lett. 2009, 9, 3290-3295. 8 Pisliakov, A.V.; Cao, J.; Kamerlin, S.C.L.; Warshel, A. Enzyme Millisecond Conformational Dynamics Do Not Catalyze the Chemical Step. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 17359-17364. 9 Cao, J. Chem. Event-Averaged Measurements of Single-Molecule Kinetics. Phys. Lett. 2000, 327, 38-44. 10 Yang, S.; Cao, J. J. Direct Measurements of Memory Effects in Single-Molecule Kinetics. Chem. Phys. 2002, 117, 10996-11009. 11 Stirnemanna, G.; Kang, S.; Zhou, R.; Berne, B. J. How Force Unfolding Differs from Chemical Denaturation. Proc. Natl. Acad. Sci. USA 2014, 111, 3413-3418. 12 Zhou, R. Berne, B. J.; Germain, R. The Free Energy Landscape for Beta Hairpin Folding in Explicit Water. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 14931-14936. 13 An, U.; Mukherjee, S. Reversibility in Protein Folding: Eeffect of B-Cyclodextrin on Covine Serum Albumin Unfolded by Sodium Dodecyl Sulphate. Phys. Chem. Chem. Phys. 2013, 15, 9375-9383. 14 Lipman, E. A.; Schuler, B.; Bakajin, O.; Eaton, W. A. Single-Molecule Measurement of Protein Folding Kinetics. Science 2003, 301, 1233-1235. 110 15 Dutta, P.; Sen, P.; Halder, A.; Mukherjee, S.; Sen, S., Bhattacharyya, K. Solvation Dynamics in a Protein–Surfactant Complex. Chem. Phys. Lett. 2003, 377, 229-235. 16 Guha, S.; Sahu, K.; Roy, D.; Mondal, S.K.; Roy, S.; Bhattacharyya, K. Slow Solvation Dynamics at the Active Site of an Enzyme: Implications for Catalysis. Biochemistry 2005, 44, 8940-8947. 17 Dickson, R.M.; Cubitt, A.B.; Tsien, R.Y.; Moerner, W.E. On/off Blinking and Switching Behaviour of Single Molecules of Green Fluorescent Protein. Nature 1997, 388, 355-358. 18 Garcia-Viloca, M.; Gao, J.; Karplus M.; Truhlar, D. G. How Enzymes Work: Analysis by Modern Rate Theory and Computer Simulations. Science 2004, 303, 186-195. 19 Antikainen, N.M.; Smiley, R.D.; Benkovic, S.J.; Hammes, G.G. Conformation Coupled Enzyme Catalysis: Single-Molecule and Transient Kinetics Investigation of Dihydrofolate Reductase. Biochemistry 2005, 44, 16835–16843. 20 Hammes, G.G. Multiple Conformational Changes in Enzyme Catalysis. Biochemistry 2002, 41, 8221-8228. 21 Margolin, G.; Barkai, E. Single-Molecule Chemical Reactions: Reexamination of the Kramers Approach. Phys. Rev. E 2005, 72, 025101. 22 Lu, H. P., Xun, L., Xie, X. S. Single-Molecule Enzymatic Dynamics. Science, 1998, 282, 1877-1882. 23 Lomholt M.A.; Urbakh M.; Metzler R.; Klafter J. Manipulating Single Enzymes by an External Harmonic Force. Phys. Rev. Lett. 2007, 98, 168302. 111 24 Flomenbom, O.; Velonia ,K.; Loos, D.; Masuo,S.; Cotlet,M.; Engelborghs,Y.; Hofkens,J.; Rowan, A.E.; Nolte, R.J.M.; der Auweraer, M.V.; de Schryver, F.C.; Klafter, J. Stretched Exponential Decay and Correlations in the Catalytic Activity of Fluctuating Single Lipase Molecules. Proc. Natl. Acad. Sci. U.S.A., 2005, 102, 2368-2372. 25 English, B.P.; Min, W.; van Oijen, A.M.; Lee, K.T. Luo, G.; Sun, H.; Cherayil, .J.; Kou, S.C.; Xie X.S. Ever-Fluctuating Single Enzyme Molecules: Michaelis-Menten Equation Revisited. Nat. Chem. Biol. 2006, 2, 87-94. 26 Lu, H. P. Probing Single-Molecule Protein Conformational Dynamics. Acc. Chem. Res. 2005, 38, 557-565. 27 Ha, T. J.; Ting, A. Y.; Liang, J.; Caldwell, W. B.; Deniz, A. A.; Chemla, D. S.; Schultz, P. G.; Weiss, S. Single-Molecule Fluorescence Spectroscopy of Enzyme Conformational Dynamics and Cleavage Mechanism. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 893-898. 28 Svoboda, K., Mitra, P. and Block, S.M. Fluctuation Analysis of Motor Protein Movement and Single Enzyme Kinetics. Proc. Natl. Acad. Sci. U.S.A. 1994, 91, 11782-11786. 29 Lu, H.P. Revealing Time Bunching Effect in Single-Molecule Conformational Dynamics. Phys. Chem. Chem. Phys. 2011, 13, 6734-6749. 30 He, Y.; Li, Y.; Mukherjee, S.; Wu, Y.; Yan, H.; Lu, H. P. Probing Single-Molecule Enzyme Active-Site Conformational State Intermittent Coherence. J. Am. Chem. Soc. 2011, 133, 14389-14395. 31 Lu, H. P. Single Molecule Spectroscopy in Chemistry, Physics and Biology: Nobel Symposium Springer, 2010, 471-494. 112 32 Gorris, H. H.; Walt, D. R. Mechanistic Aspects of Horseradish Peroxidase Elucidated through Single-Molecule Studies. J. Am. Chem. Soc.2009, 131, 6277-6282. 33 Hassler, K.; Rigler, P.; Blom, H.; Rigler, R.; Widengren, J.; Lasser, T. Dynamic Disorder in Horseradish Peroxidase Observed with Total Internal Reflection Fluorescence Correlation Spectroscopy. Opt. Express. 2007, 15, 5366-5375. 34 Edman, L.; Foldes-Papp, Z.; Wennmalm, S.; Rigler, R. The Fluctuating Enzyme: a Single Molecule Approach. Chem. Phys. 1999, 247, 11-22. 35 Klibanov, A.M. Improving Enzymes by Using Them in Organic Solvents. Nature 2001, 409, 241-246. 36 Fersht, A. R. Structure and Mechanism in Protein Science: a Guide to Enzyme Catalysis and Protein Folding, Freeman publishing, 1999. 37 Fersht, A. R.; Matouschek, A.; Serrano, L. The Folding of an Enzyme. I. Theory of Protein Engineering Analysis of Stability and Pathway of Protein Folding. J.Mol. Biol. 1992, 224(3), 771-782. 38 Liu, R. C.; Garcia-Manyes, S.; Sarkar, A.; Badilla, C. L.; Fernandez, J. M. Mechanical Characterization of Protein L in the Low-Force Regime by Electromagnetic Tweezers/Evanescent Nanometry.Biophys. J. 2009, 96, 3810-3821. 39 del Rio, A.; Perez-Jimenez, R.; Liu, R. C.; Roca-Cusachs, P.; Fernandez, J. M.; Sheetz, M. P. Stretching Single Talin Rod Molecules Activates Vinculin Binding. Science 2009, 323, 638-641. 113 40 Zhou, H.X.; Wlodek, S.T.; McCammon J.A. Conformation Gating as a Mechanism for Enzyme Specificity. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 9280-9383. 41 Hanson, J.A.; Duderstadt, K.; Watkins, L.K.; Bhattacharyya, S.; Brokaw, J.; Chu, J.W.; Yang, H. Illuminating the Mechanistic Roles of Enzyme Conformational Dynamics. Proc. Natl. Acad. Sci. USA 2007, 104, 18055-18060. 42 Onuchic, J.N.; Wolynes, P.G. Theory of Protein Folding. Curr. Opin. Struc. Biol. 2004, 14, 70-75. 43 Wright, P.E.; Dyson, H.J. Linking Folding and Binding. Curr. Opin. Struc. Biol. 2009, 19, 31-38. 44 Korkegian, A.; Black, M.E.; Baker, D.; Stoddard, B.L. Computational Thermostabilization of an Enzyme. Science 2005, 308, 857-860. 45 Li, H.; Fernandez, J.M. Force-Clamp Spectroscopy Monitors the Folding Trajectory of a Single Protein. Science 2004, 303,1674-1678. 46 Carrion-Vazquez, M.; Oberhauser, A. F.; Fowler, S. B.; Marszalek, P. E.; Broedel, S. E.; Clarke, J.; Fernandez, J. M. Mechanical and Chemical Unfolding of a Single Protein: A Comparison. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 3694-3699. 47 Kienbergera, F.; Kadaa, G.; Muellerb, H.; Hinterdorfera, P. J. Single Molecule Studies of Antibody-Antigen Interaction Strength Versus Intra-Molecular Antigen Stability. Mol. Biol. 2005, 347, 597–606. 114 48 Nevo, R.; Stroh, C.; Kienberger, F.; Kaftan, D.; Brumfeld, V.; Elbaum, M.; Reich, Z.; Hinterdorfer, P. A Molecular Switch Between Alternative Conformational States in the Complex of Ran and Importin Bold Beta1. Nature Struct. Biol. 2003, 10, 553 – 557. 49 Stigler, J.; Reif, M. Calcium-Dependent Folding of Single Calmodulin Molecules. Proc. Natl. Acad. Sci. U.S.A., 2012, 109, 17814–17819. 50 Stigler, J.; Ziegler, F.; Gieseke, A.; Gebhardt. C.M.; Reif, M. The Complex Folding Network of Single Calmodulin Molecules. Science 2011, 334, 512-516. 51 Shank, E.A.; Cecconi, C.; Dill, J.W.; Marqusee, S.; Bustamante, C. The Folding Cooperativity of a Protein is Controlled by Its Chain Topology. Nature 2010, 465, 637–640. 52 Guo, Q.; He, Y.; Lu, H. P. Manipulating and Probing Enzymatic Conformational Fluctuations and Enzyme-Substrate Interactions by Single-Molecule FRET-Magnetic Tweezers Microscopy. Phys. Chem. Chem. Phys. 2014, 16, 13052-13058. 53 Åqvist, J.; Warshel , A. Simulation of Enzyme Reactions Using Valence Bond Force Fields and Other Hybrid Quantum/Classical Approaches. Chemical Rev. 1993, 93, 2523-2544. 54 Garcia-Viloca, M.; Gao, J.; Karplus M.; Truhlar, D. G. How Enzymes Work: Analysis by Modern Rate Theory and Computer Simulations. Science 2004, 303, 186-195. 55 Prakash, M.K.; Marcus R.A. An Interpretation of Fluctuations in Enzyme Catalysis Rate, Spectral Diffusion, and Radiative Component of Lifetimes in Terms of Electric Field Fluctuations. Proc. Natl. Acad. Sci. U.S.A., 2007, 104, 15982–15987. 56 Strick, T. R.; Allemand, J. F.; Bensimon, D.; Bensimon, A.; Croquette, V. The Elasticity of a Single Supercoiled DNA Molecule Science 1996, 271, 1835-1837. 115 57 Gosse, C.; Croquette, V. Magnetic Tweezers: Micromanipulation and Force Measurement at the Molecular Level. Biophys. J. 2002, 82, 3314--3329. 58 Smith, S. B.; Finzi, L.; Bustamante, C. Direct Mechanical Measurements of the Elasticity of Single DNA Molecules by Using Magnetic Beads. Science 1992, 258, 1122-1126. 59 Haber, C.; Wirtz, D. Magnetic Tweezers for DNA Micromanipulation Rev. Sci Instrum. 2000, 71, 4561-4570. 60 Phillips, JC et al (2005) Scalable Molecular Dynamics with NAMD. J.Comput. Chem., 26:1781-1802, 2005. 61 Tanase, M.; Biais, N.; Sheetz, M. In Cell Mechanics; Wang, Y. L., Discher, D. E., Eds. 2007; Vol. 83, p 473-493. 62 Kollmannsberger, P.; Fabry, B. High-Force Magnetic Tweezers with Force Feedback for Biological Applications. Rev. Sci Instrum. 2007, 78:114301. 63 Kruithof, M.; Chien, F.; de Jager, M.; van Noort, J. Subpiconewton Dynamic Force Spectroscopy Using Magnetic Tweezers. Biophys. J. 2008, 94, 2343-2348. 64 Leuba, S. H.; Karymov, M. A.; Tomschik, M.; Ramjit, R.; Smith, P.; Zlatanova, J. Assembly of Single Chromatin Fibers Depends on the Tension in the DNA Molecule: Magnetic Tweezers Study. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 495-500. 66 Ribeck, N.; Saleh, O. A. Multiplexed Single-Molecule Measurements with Magnetic Tweezers. Rev. Sci Instrum. 2008, 79: 094301. 116 66 Yan, J.; Skoko, D.; Marko, J. F. Near-Field-Magnetic-Tweezer Manipulation of Single DNA Molecules. Phys. Rev. E 2004, 70:011905. 67 Yanagida, T.; Ishii, Y. Single Molecule Dynamics in Life Science, Press: Wiley, 2008. 68 Bustamante, C.; Chemla, Y.R. ; Moffitt, J.R. Single Molecule Techniques. Cold Spring Harbor Laboratory Press, 2008; 297-325. 69 Siuti P.; Retterer S.T.; Choi C.K.; Doktycz M.J. Enzyme Reactions in Nanoporous, Picoliter Volume Containers. Anal. Chem. 2012, 84, 1092-1097. 70 Sullivan, C. J.; Venkataraman, S.; Retterer, S. T.; Allison, D. P.; Doktycz, M. J. Comparison of the Indentation and Elasticity of E. coli and its Spheroplasts by AFM. Ultramicroscopy, 2007, 107, 934-932. 71 Dyson, H.J.; Wright, P.E. Intrinsically Unstructured Proteins and Their Functions. Nat Rev Mol Cell Biol.2005, 6, 197-208. 72 Tompa, P. Intrinsically Unstructured Proteins. Trends Biochem Sci. 2002, 10, 527-533. 73 Dunker A.K.; Lawson J.D.; Brown C.J.; Williams R.M.; Romero P.; Oh J.S.; Oldfield C.J.; Campen A.M.; Ratliff C.M.; Hipps K.W.; Ausio J.; Nissen M.S.; Reeves R.; Kang C.; Kissinger C.R.; Bailey R.W.; Griswold M.D.; Chiu W.; Garner E.C.; Obradovic Z. J Intrinsically Disordered Protein. Mol Graph Model. 2001, 1, 26-59. 74 Sugase, K.; Dyson, H.J.; Wright, P.E. Mechanism of Coupled Folding and Binding of an Intrinsically Disordered Protein. Nature 2007, 447, 1021-1025. 117 75 Tran H.T.; Mao, A.; Pappu, R.V. Role of Backbone-Solvent Interactions in Determining Conformational Equilibria of Intrinsically Disordered Proteins. J. Am. Chem. Soc. 2008, 130, 7380-7392. 76 Eliezer, D. Biophysical Characterization of Intrinsically Disordered Proteins. Curr. Opin. Struc. Biol. 2009, 19, 23-30. 77 Elam, W.A.; Schrank, T.P.; Campagnolo, A.J.; Hisler, V.J. Evolutionary Conservation of the Polyproline II Conformation Surrounding Intrinsically Disordered Phosphorylation Sites. Protein Science, 2013, 22, 405-417. 78 Ferreom, A.C.M.; Gambin, Y.; Lemke, E.A.; Deniz, A.A. Interplay of α-Synuclein Binding and Conformational Switching Probed by Single-Molecule Fluorescence. Proc. Natl. Acad. Sci. U.S.A.2009, 106, 5645-5650. 79 Wells, M.; Tidow, H.; Rutherford, T.J.; Markwick, P.; Jensen, M.R.; Mylonas, E.; Svergun, D.I.; Blackledge, M.; Fersht, A.R. Structure of Tumor Suppressor p53 and Its Intrinsically Disordered N-Terminal Transactivation Domain. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 5762–5767. 80 Reichmann, D.; Xu, Y.; Cremers, C.M.; Ilbert, M.; Mittelman,R.; Fitzgerald, M.C.; Jakob, U. Order out of Disorder: Working Cycle of an Intrinsically Unfolded Chaperone. Cell, 2012, 148, 947-957. 81 Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Intrinsically Disordered Proteins in Human Diseases: Introducing the D2 Concept. Annu. Rev. Biophys. 2008, 37, 215–246. 118 82 Dyson, H.J.; Wright, P.E. Coupling of Folding and Binding for Unstructured Proteins. Curr. Opin. Struc. Biol. 2002, 12, 54-60. 83 Dyson, H.J.; Wright, P.E. Linking Folding and Binding. Curr. Opin. Struc. Biol. 2009, 19, 31-38. 84 Wang, Y.; Chu, X.; Longhi, S.; Roche, P.; Han, W.; Wang, E.; Wang, J. Multiscaled Exploration of Coupled Folding and Binding of an Intrinsically Disordered Molecular Recognition Element in Measles virus Nucleoprotein. Proc. Natl. Acad. Sci. U.S.A.2013, 110, E3743–3752. 85 Viani, M. B.; Pietrasanta, L. I.; Thompson, J. B.; Chand, A.; Gebeshuber, I. C.; Kindt, J. H.; Richter, M.; Hansma, H. G.; Hansma, P. K. Probing Protein−Protein Interactions in Real Time. Nat. Struct. Biol. 2000, 7, 644-647. 86 Ganguly, D.; Chen, J.H. Topology-Based Modeling of Intrinsically Disordered Proteins. Proteins: Struct., Funct., Bioinf. 2011, 79, 1251-1266. 87 Venkitakrishnan,R.P.; Zaborowski,E.; McElheny,D.; Benkovic, S.J.; Dyson, H.J.; Wright, P.E. Conformational Changes in the Active Site Loops of Dihydrofolate Reductase During the Catalytic Cycle. Biochemistry 2004, 43, 16046-16055. 88 We note that on a given HRP molecule, biotin or DMS are able to covalently link to lysine residue in the amino acid sequence, which results in protein immobilization complexity that a few different tethering conditions are possible for HRP protein molecules when linking to coverslip or magnetic beads. However, in our single-molecule TIRF measurement, we compare activity change of each HRP molecules individually under different conditions that with and 119 without magnetic pulling force applied. As a result, although we did not necessarily pinpoint one specific lysine residue pair on protein molecules for tethering, our observation of different enzymatic activity associated with various enzyme conformational manipulation conditions are systematic and well-defined. 120 CHAPTER V. STEERED MOLECULAR DYNAMICS SIMULATION STUDIES OF THE CONFORMATIONALLY DEFORMED ENZYMES MANIPULATED BY SINGLE MOLECULE MAGNETIC TWEEZERS 5.1. Introduction Steered Molecular Dynamics (SMD) simulation, sometimes also being named as force probe simulation, is originally designed for the purpose of applying external force onto a protein molecule to pull the protein along desired directions. In our experiments discussed in chapter III and chapter IV, we apply force on protein by immobilize a single protein at its one given residue position, while pulling at another residue position which is tethered covalently to magnetic bead. Such force that we apply to manipulate protein structure allows us to pull a single protein molecule along a given direction, or a given degree of freedom at atomic level. SMD simulation is ideal for this type of scenario at molecular level. The SMD simulation is performed using the NAMD software package, developed by Schulten’s group. Typically, there are two different types of protocols for simulation by SMD: one in which the target molecule is pulled at constant velocity, and one in which the target molecule is pulled with constant force. Although the constant-force simulation will fit the scenario of our experiments better, considering the computer facility and our goal which focused on revealing the active domain structural information when the protein is conformationally deformed, we chose the constant-velocity type method to carry out our SMD simulation. Details will be discussed in later sections. 121 5.2. Estimating conformational stretching extent from HPPK simulation In chapter III, we have used FRET-Magnetic Tweezers Microscopy to manipulate the conformation of a single HPPK protein molecule. From the FRET measurement results, we can clearly see that single-molecule HPPK enzyme is stretched out in conformation under the external pulling force. And such conformational manipulation has been demonstrated for its high reproducibility and feasibility of the force manipulation of the conformational changes of the single HPPK protein molecule via magnetic tweezers. However, since FRET spectroscopy only reports distance changes between two specific residue positions where dye indicators are labeled on a protein molecule, what we can really obtain from FRET data is a projection of the molecular conformational change on the FRET donor-FRET acceptor direction. Such information is informative for the purpose of dynamical analysis such as conformational fluctuation rate of protein molecules, yet it does not show atomic details of protein structure with external mechanical impact is applied on target sample protein molecules. In this section, we will study what impact such conformational manipulation will give to HPPK protein structure by Steered Molecular Dynamics (SMD) simulation. As discussed in chapter 3.2.2, we have labeled dye molecule Cy3 at residue position 48 and Cy5 on residue position 151 on HPPK protein molecule, as shown in figure 5.1A and 5.1B. A combined magnetic tweezers and simultaneous FRET spectroscopic microscopy is used to apply and to monitor conformational manipulation on single HPPK molecule. We observed that when a single HPPK protein molecule is pulled by our magnetic tweezers, the mean value of FRET efficiency between Cy3 and Cy5 shifted from 0.5 to 0.3, indicating the single HPPK molecule is stretched out in conformation leading to an extension between 4Å to 6Å in the distance between Cy3 and Cy5 residue position, as shown in figure 5.1C. Details have been published in our 122 previous paper.1 To find out how much a protein molecule is stretched while being pulled by magnetic tweezers in experiment, we performed a steered molecular dynamic (SMD) simulation of HPPK protein molecule. As we have discussed in chapter 5.1, there are two types of SMD simulations: constant-force pulling simulation and constant-speed pulling simulation. In our steered MD simulation, we did constant speed pulling simulation to illustrate the conformational distortion of HPPK protein molecule responding to external stretching force. There are two reasons for this choice: (a) the force we apply in experiment is in such a fine scale that calculation time would be too long for constant force simulation; (b) In chapter III, we have already demonstrated the capability of the force generated by our magnetic tweezers set to stretch the conformation of a single protein molecule. The initial coordinates of HPPK were obtained from Protein Data Bank (PDB code 1HKA), set in aqueous environment during simulation, with periodic boundary condition set for a rectangular shape water box with 67.9Å in length, 54.7Å in width and 67.3Å in height. MD simulation is performed using program NAMD, version 2.9. Protein molecule is set in water solvation condition under CHARMM type force field (par_all27_prot_lipid.inp). Boundary condition is applied for a time step of 1 fs. Considering the computation time, we set the pulling speed as 0.5 Å/ps.1-3 Constant temperature at 293 K during the simulation is maintained by Langevin thermostat, with Langevin damping coefficient set at 1 ps-1. Constant pressure is maintained at 1atm using Langevin piston. Non-bonded interactions were calculated using particle mesh Ewald (PME) full electrostatics; cutoff of the van der Waals energy was set at 12.0 Å, with switch distance at 10.0 Å and pair-list distance set at 13.5 Å. PME grid spacing is set at 1.0 Å. We have also run 20 times the HPPK molecule under same conditions but without any 123 pulling events to test the HPPK conformational thermal fluctuation at its equilibrium condition as a controlling group test. The results are shown in figure 5.1D and 5.1E. Figure 5.1D shows that for a single HPPK molecule in aqueous environment without any perturbation applied on it, the distance between residue 48 and 151 displays a fluctuation within 2Å induced by thermal motion. Figure 5.1E shows that upon being stretched, this distance will be distorted, which is 4Å to 6Å from FRET measurement in our previous paper. One point needs mentioning was there are 5 lysine residues in an HPPK protein molecule as possible tethering positions: residues 23, 85, 119, 154 and 157. Two out of these five lysine residues participate in the conformational manipulation: for each single HPPK protein molecule, it is immobilized on coverslip through one lysine residue, and being tethered to magnetic beads via another lysine residue. Since the conformational stretching by magnetic tweezers has to be achieved through applied force on magnetic beads, in magnetic tweezers pulling experiment, the HPPK molecule will be stretched in the direction formed by two out of these five lysine residues. For residue pair 85-154, it will need to be stretched for 17Å to 18Å to make the distance between residues 48-151 extend for 4Å to 6Å, while stretching through any other possible lysine residue pair on HPPK molecule requires significantly larger distortion to achieve similar effect. In other words, the HPPK protein will be stretched no less than for 17Å to 18Å when FRET shift from 0.5 to 0.3 is observed. Therefore, in HRP simulation, we also assumed that our HRP molecule is stretched 17Å to 18Å when being pulled by magnetic tweezers. 124 Fig. 5.1. SMD simulation of HPPK molecule pulling by magnetic tweezers. The magnetic tweezers pulling experiment is described in our previous publication.1 (A) Natural HPPK protein molecule. (B) HPPK protein molecule being pulled by magnetic tweezers. (C) FRET efficiency distributions of single HPPK molecules under pulling (Red) and releasing (Blue) manipulation. (D) Distortion of distance between residue 48 and 151 induced by thermal fluctuation of HPPK protein molecule. (E) Distortion of distance between residue 48 and 151 induced by pulling force applied on HPPK protein molecule. 125 5.3. SMD simulation study of HRP protein molecule 5.3.1. SMD simulation of HRP protein molecule in one tethering condition In chapter III, we have achieved conformational manipulation of a single HPPK protein molecule using magnetic tweezers. Based on this demonstration of capability that our magnetic tweezers is able to apply pico-newton scale force on single protein molecule, we can start to study the impact of protein function from such fine-scale conformational manipulation. The first choice will be testing the impact of such applied force on HPPK protein function. Unfortunately, the reaction that HPPK serve as enzyme does not release fluorescence product available for detection in our lab. Hence we chose another protein, HRP, instead, as we have discussed in chapter IV. On the other hand, for FRET measurement, labelling dye molecules on protein requires mutation to introduce cysteine residues. Yet for HRP molecules, mutation on cysteine residue will significantly affect its conformation leading to change in its function; not to mention that there are 6 cysteine residues on an HRP molecule, making it hard to do selective mutation. As a result, we take a compromise to estimate the stretching extent of protein molecule under magnetic tweezers manipulation from our previous HPPK-FRET study. To quantitatively understand the impact of structural distortion on protein function, we performed a steered molecular dynamic (SMD) simulation to help our understanding on how the conformational manipulation by magnetic tweezers affects the active domain on a protein molecule.4, 5 From the single-molecule FRET measurement result in our recent publication, we can identify that a single protein molecule is typically stretched no less than 18Å using our magnetic tweezers under the same experimental conditions reported (see supporting information for details).6 For HRP protein molecule, since HRP can only be 126 linked to modified glass or tethered to magnetic beads through lysine residue, we set lysine residue 65 and lysine residue 174 on HRP protein molecule stretched 17Å as an example to discover the corresponding active site conformational change. As shown in Figure 6, we use the distance between residue 68 and residue 178 to characterize conformational distortion on active site of HRP protein molecule. In the steered MD simulation, we did constant speed pulling simulation to illustrate the conformational distortion of HRP protein molecule responding to external stretching force. Similarly to SMD simulation of HPPK in section 5.2, here the HRP protein molecules are stretched in the same pulling condition by the same single molecule fluorescence magnetic tweezers microscopic approach. The goal for this simulation is to characterize the distortion scale of the active site of a HRP protein once the HRP protein molecules are in the same stretching condition via our magnetic tweezers, compared with the results from our previous published work.36 The initial coordinate of HRP protein molecule is taken from Protein Data Bank (PDB code 1W4Y). MD simulation is performed using program NAMD, version 2.9. Protein molecule is set in water solvation condition under CHARMM type force field (par_all27_prot_lipid.inp). Boundary condition is applied for a time step of 1 fs. Considered the computation time, we set the pulling speed as 0.5 Å/ps.1-3 Constant temperature at 293 K during the simulation is maintained by Langevin thermostat, with Langevin damping coefficient set at 1 ps-1. Constant pressure is maintained at 1atm using Langevin piston. Non-bond interaction is calculated using particle mesh Ewald (PME) full electrostatics; cutoff of the van der Waals energy is set at 12.0 Å, with switch distance at 10.0 Å and pair-list distance set at 13.5 Å. PME grid spacing is set at 1.0 Å. We have also run 20 times MD simulation of HRP molecule at same 127 condition without any pulling events to test the HRP conformational thermal fluctuation at its equilibrium condition as a controlling group test. 128 Figure 5.2. SMD simulation results show the scheme of the distortion of active site when the protein is pulling by magnetic tweezers. (A) Natural HRP protein molecule. (B) HRP protein molecule being pulled by magnetic tweezers. (C) Distortion of distance between residue 68 and 178 induced by thermal fluctuation of HRP protein molecule. (D) Distortion of distance between residue 68 and 178 induced by pulling force applied on HRP protein molecule. (E)(G)(I) Projection on Cartesian coordinate of the distance distortion between residue 68 and residue 178 for HRP protein induced by thermal fluctuation in unperturbed condition. (F)(H)(J) Projection on Cartesian coordinate of distance distortion between residue 68 and residue 178 for HRP protein in stretched condition. As shown in Figure 5.2A, the initial condition of the HRP protein molecule is under equilibrium conformation in aqueous environment without the force pulling perturbation applied. In about 20 independent simulation events, the distance between residue 68 and residue 178 is observed to have a fluctuation within 2Å due to conformational thermal fluctuation of HRP protein, as shown in Figure 5.2C, while the corresponding projections on spatial Cartesian coordinate are shown in Figure 5.2E, 5.2G and 5.2I. Figure 5.2B illustrates how the distance of the residue pair 68-178 gets distorted when the HRP molecule is stretched in experiment by magnetic tweezers, indicating distortion of active site in HRP protein. Figure 5.2D shows the statistical results from 20 simulation events that the distance extension of residue 68 and residue 178 gets extended for about 8-10Å when the experimentally tethered lysine residue 65 and residue 174 on protein molecule are stretched for 17Å. The corresponding projections on spatial Cartesian coordinate are shown in Figure 5.2F, 5.2H and 5.2J. Such responses from SMD simulation conceptually reveal that in our 129 experiment condition, when being stretched by magnetic tweezers, the active site on HRP protein molecule is distorted in an extent that significantly beyond its thermal conformational fluctuation. We also note here that because HRP protein in experiment is linking to either coverslip or magnetic bead via connection with lysine. Therefore, similar to HPPK case, there are multiple possible tethering conditions of the protein molecule to the coverslip or magnetic beads, leading to multiple possible stretching types for different HRP protein molecules too. The active site distortion response shown in Figure 5.2 is from one possible pattern to stretch HRP molecules. Hence, we tested all the possible stretching condition for HRP protein molecule to reveal that conformational distortion beyond thermal fluctuation range of active site occurs for most stretching types for HRP molecule, which will be discussed in section 5.3.2. 5.3.2. SMD study on all possible stretching type of HRP protein molecule There are 6 lysine residues in the HRP amino acid sequence. As being discussed earlier, HRP molecule can only be linked to modified glass or tethered to magnetic beads through lysine residue, and in experiment the HRP protein can only be stretched through direction formed by two out of these lysine residues. From mathematical combination, there are 15 possible lysine pair combination, leading to 15 different possible stretching types for different HRP protein molecules. We studied all these 15 possible stretching situations by SMD simulation, as shown in figure 5.3. We set three residue pairs to illustrate distortion on the active domain of HRP molecule: residue pair 68-178, residue pair 140-228 and residue pair 30-252, as shown in figure 5.3A. The SMD environment parameters are all the same as described in materials and methods 130 section. From the sim mulation results shown in i figure 5.3B B, 5.3C andd 5.3D, we find that whenn an HRP mollecule is streetched by maagnetic tweeezers, its actiive domain w will have disstortion significan ntly greater than t thermall conformatiional fluctuaation for mosst of the possible stretchhing types. ue pairs to illlustrate disto ortion on thee active dom main when ann HRP molecule Fig. 5.3. Three residu hed by magneetic tweezers. All the bllack dashed line are therrmal fluctuattion range off is stretch selected residue r pairss, obtained from f statisticcal result fro m 20 simulaation events of HRP prottein in aqueou us equilibriu um condition n. Blue dash hed lines are doubled rannge of black ones, set as stricter th hreshold. (A A) Scheme of the three seelected residdue pairs. (B B) Distortion of distance between residue 68 and a residue 178 1 in all 15 possible strretching condditions for H HRP moleculle. ortion of disttance betweeen residue 14 40 and residdue 228 in alll 15 possiblee stretching (C) Disto condition ns for HRP molecule. m (D D) Distortion n of distance between ressidue 30 andd residue 2522 in all 15 possible stretch hing conditio ons for HRP P molecule. 131 5.3.3. Disstortion in unfolding u sim mulation To T quantitativ vely comparre difference in impact o n active dom main betweenn our experimeental stretchiing condition n and unfold ding conditioon for HRP pprotein moleecule, we tesst the active sitte conformattional distorttion in largerr unfolding ssituation by simulation. We still sellect the three residue pairr 68-178, 140 0-228 and 30 0-252 to chaaracterize coonformationaal change of hown in figu ure S3A and S3B, when the distancee between lyssine active sitte of HRP prrotein. As sh residue 65 6 and residu ue 174 is streetched rough hly 50Å, the distance bettween residuue 68 and 1778 is extended d for 38Å, the distance beetween resid due 140 and 228 is extennded for 14Å Å, while the distance between residue 30 and 252 decreassed for 1Å. S Such active site distortioons show thaat the conformaational distortion achieved in our maagnetic tweeezers experim ment is signiificantly smaaller from protein unfoldin ng, indicatin ng that the co onformationaal distortion is more likeely to be on tertiary structure rath her than protein unfoldin ng, which is iin accordancce with our pprevious studdy. 1 onal distortio on in larger uunfolding sittuation. Thee initial Fig. 5.4. Active site conformatio A) coordinatte of HRP prrotein moleccule is taken from Proteiin Data Bankk (PDB codee 1W4Y). (A 132 Equilibrium conformation of HRP protein in aqueous solution. (B) Conformation of HRP protein when its lysine residue 65 and 174 is stretched for 48.8Å. 5.4. References 1 Gao, M.; Wilmanns, M.; Schulten, K. Steered Molecular Dynamics Studies of Titin I1 Domain Unfolding. Biophys. J. 2002, 83: 3435-3445. 2 Lu H, Isralewitz, B.; Krammer, A.; Vogel, V.; Schulten, K. Unfolding of Titin Immunoglobulin Domains by Steered Molecular Dynamics Simulation. Biophys.1998, J. 75: 662-671. 3 Gräter, F., Shen, J.; Jiang, H.; Gautel, M.; Grubmüller, H. Mechanically Induced Titin Kinase Activation Studied by Force-Probe Molecular Dynamics Simulations. Biophys. J.2005, 88: 790-804. 4 NAMD was developed by the Theoretical and Computational Biophysics Group in the Beckman Institute for Ad-vanced Science and Technology at the University of Illinois at Urbana-Champaign. 5 Phillips J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable Molecular Dynamics with NAMD. J.Comput. Chem.,2005, 26:1781-1802, 2005. 6 Guo, Q.; He, Y.; Lu, H.P. Manipulating and Probing Enzymatic Conformational Fluctuations and Enzyme-Substrate Interactions by Single-Molecule FRET-Magnetic Tweezers Microscopy. Phys. Chem. Chem. Phys. 2014, 16: 13052-13058. 133 CHAPTER VI. DESIGN AND IMPLEMENTATION OF A QUADRUPOLE MAGNETIC TWEEZERS In this chapter, we discussed a newly developed technical improvement for higher controlling ability for next generation of magnetic tweezers. 6.1 History of Instrumental Design for Magnetic Tweezers Magnetic tweezers has been widely acknowledged for its capability to apply pN scale force on multiple biological specimens and single molecules simultaneously, as we have already discussed in chapter II. It is from 1996 that the word ‘magnetic tweezers’ started to be widely used for one certain type of experiment using magnetic field to apply external force onto single molecule, especially DNA or protein molecule, through paramagnetic nanoparticles tethered on those molecules. For example, Bensimon and Croquette have used magnetic tweezers to study DNA topological properties since 1996;1 Ingber’s group has made a lot of achievements using magnetic tweezers for transportation in living cell condition;2‐3 Fernandez and Sheetz has developed magnetic tweezers as a force spectroscopy method to study protein unfolding problems, etc.4 Scientific aim of magnetic tweezers when being used as a force approach to apply mechanical manipulation.in biophysics studies focuses on two aspects. The first one is using magnetic tweezers to manipulate target molecules such as DNA or protein to study their roles in biological processes.5‐6 The second aspect is using magnetic tweezers to manipulate paramagnetic beads for the purpose of drug delivery or cell mechanics applications.7‐8 Therefore, versatile force to be applied on target specimen and precise manipulation of magnetic beads has been a central topic in the field of magnetic tweezers for a long time. 134 In practical, there are three technical requirements for magnetic tweezers to be successfully applied in biophysical experiments, especially single molecule biophysical research. Firstly it needs to be able to achieve force ranged from sub-pN to nN, depending on specific sample system experimental measurements: for molecule manipulation purpose, such as single molecule studies of DNA or protein molecules, force range from sub-pN to 100 pN are always preferred, while for the purpose of transportation of magnetic particles in biological specimen, larger force from a few hundreds of pN to even nN is necessary. Secondly, since magnetic tweezers is a force approach without the capability to provide spatial information of target sample molecules, a correlated optical observation method is always needed to provide spatial information of the sample system simultaneously, which requires that the setup of magnetic tweezers needs to be not too large to be launched with correlated optical observation instruments. Thirdly, since the core part of magnetic tweezers is always the magnet which generating magnetic fired, for the purpose of the capability to move magnetic beads in arbitrary directions, a classical design of the magnet part is multi-channel magnetic tweezers, which applies multidimensional force with several magnetic poles. Since 1996, many research works has been made to develop various types of magnetic tweezers aiming at these requirements. A two-pole electromagnet combination has been developed back in 1992 by Gore’s group to generate large force onto paramagnetic metal particles with 10 µm diameter for biological tissue studies.9 In 1996, a computer controlled fourpole magnetic manipulator that capable to generate force at pN scale onto magnetic beads in 2.8 µm size has been developed by Leiber’s group.10 The force scale it can achieve at that time is already ideal for single molecule experiments. However, the design of multidimensional magnet setup in 1990s was always very space consuming, limiting technical developments from being 135 directly benefit scientific research. For example, scientists had to rely on simple one piece of permanent magnet to apply force onto magnetic beads for cell-substrate studies in 1998.11 After 2000s, some scientists started to focus their effort on designing magnetic tweezers setup that small enough to be correlated into single molecule measurements in experiment, while still being able to generate force at pN scale, which is always required for molecule level manipulation. In 2000, a smaller, yet still complicated design of two-pole electromagnet coil was developed by Habor and Wirtz.12 In 2002, Croquette’s group designed a six-coiled electromagnet with 2cm in diameter, which is small enough to be launched above a normal inverted microscope, yet just generating twisting force specifically for DNA topological research.13 In 2003, a computer controlled two-pole electromagnet setup in a compactible size that capable to be fitted onto the stage of a normal inverted microscope was developed by Forgacs’s group.14 Meanwhile, from 2000 to 2004, needle shaped electromagnet has been studied throughout by Ingber’s group.2‐3 In 2005, Fisher et al for the first time developed a four pole needle shaped electromagnet which can apply force in a wide range from a few pN to thousands of pN, yet still having a few mechanical trade-offs in its design.15 In 2006, a symmetric face-centered-cubic pole combination has been designed by Fisher et al to apply magnetic force manipulation which can be easily correlated with optical observation in experiments. Yet the force they achieved via the six-pole setup was still large from a few hundreds of pN to nN scale onto paramagnetic beads small as 1 µm size.16 In 2007, Fabry’s group developed a single needle shaped electromagnet with strong near-tip magnetic field gradient and hysteresis compensation design to achieve large force up to 10 nN.17 By 2008, Driel’s group developed a four-pole needle shaped electromagnet with even smaller size making the magnet part to be better fitted with normal inverted microscope.18 Meanwhile, there are also 136 some theoretical studies to modeling magnetic field for various types of magnets.19 However, there are still some technical limitations in each design of those multi-pole electromagnet setups in different aspects: hysteresis effect, durability of the needle shaped electromagnet, lacking of spatial freedom of the electromagnets involved in the multi-pole design, etc. As a result, for biophysical experiments, scientists still rely on the simple way to generate reliable magnetic force, although in a limited range. For example, until 2009, a few pieces of simple permanent magnets were still used for manipulation research for DNA molecules.20 6.2 6.2.1 The Multi-Channel Magnetic Tweezers An Introduction of the Multi-Dimensional Magnetic Tweezers Setup in Our Lab In our studies, we also developed a multi-channel electromagnet to apply magnetic force onto paramagnetic beads in in arbitrary directions. A conceptual figure of the design of our quadrupole magnetic tweezers is shown in figure 6.1. Four magnetic poles are launched in a quadrupole configuration on an alumni plate. Each magnetic pole is an electromagnet which is custom-made in the Instrument Development Laboratory (IDL) located in the Environmental Molecular Sciences Laboratory (EMSL) facility of Pacific Northwestern National Lab (PNNL). The electromagnet is built by twining coils on a sharpened metal rod as probe. Magnet field will be generated and sent out through the tip of the metal rod once current is applied. The coils have been tested to be able to bear voltages up to 5V before showing obvious temperature change from heating effect of applied current. In this way, the four poles can generate magnetic field and hence to apply force on paramagnetic beads in arbitrary directions in 2D plane. 137 Figure F 6.1. A conceptuall scheme of the t quadrupoole magneticc tweezers seetup. Four electromagnets poless are set on an a alumni plaate which caan be launchhed on a simpple inverted microsco ope. The tip end of each poles are cu ustom-made to be sharpeen metal to ggenerate stroong magnet flux f gradientt. The four poles p are insttalled on thee plate via foour independdent 2D micrometers, allowin ng us to adju ust physical position p of eeach magnet pole independently. Heence nce among four f metal tip ps of poles are a tunable inn a range froom touching each other tto a the distan few centiimeters. Thee controller was w used to be a joystickk with a few w limitations in its function, while currrently it hass been upgraade to a digittalized contrroller, whichh will be disccussed in secction 6.2.2. The T function of this apparratus to applly external fo force onto paaramagnetic beads has already been b tested by b Jason J. Han H and Alex x Li from W Washington Sttate Universsity in 2004. 3‐4 In brief, a few λ‐DNA A molecules are immobillized at one end on glasss coverslip inn aqueous 138 environm ment, while being b tethereed on the oth her end by m magnetic beadds with 1.5 µ µm diameterr. An additionaal permanentt magnet is needed n on th he top to helpp in obtaininng repetitive response froom the tetherred beads. In plane 2D-m manipulation n can be appplied by elecctromagnet ppoles on the setup illustrated in figure 6.1. 6 DNA molecules m sen nse the magnnetic force viia tethered bbeads. Net fforce our differentt directions has h been testted. Flash laamp has beenn used as ligght source, w while toward fo the respo onse of magn netic beads can c be observ ved from eyeepiece on thhe microscoppe. 6 Concepttual scheme of an experiiment testingg the functioon electromagnet poles. Figure 6.2. The T result of this testing experiment e is i shown in ffigure 6.3. W When applyinng mechaniccal pulling fo orce via mag gnetic field, the regulated d movementt of the magnnetic beads ccan be obserrved by a norm mal optical microscope. m The moving g pattern of m magnetic beeads can be m manipulated by changing g the directio on of externaal net total magnetic m fieldd. Firstly, thhere are no m magnetic fielld 139 applied onto o the sample system, and with thee help of addditional perm manent magnnet, Browniaan fluctuatio on of the teth hered bead is limited. Then, magnettic field towaard differentt net directioons are applieed consecutiively: firstly y, the net field is set tow ward west, theen toward eaast, afterwarrd, toward west w again; laater, net field d is set towaard southeastt direction, thhen southweest direction,, finally to oward northeeast direction n before it iss totally remooved. The teesting result of magneticc beads is shown s in fig gure 6.3B. Figure 6.3. 6 A testing g experimentt examinatio on the electroomagnet funnctions. (A) Sample systtem for the teest. (B) Expeerimental ressults show th he response oof the param magnetic beadds toward external net n magneticc field, preseenting the caapability of eelectromagneet poles to aapply force 140 manipulating movements of magnetic beads. (1) No field; (2) Net field toward West; (3) Net field toward East; (4) Net field toward West; (5) Net field toward Southeast; (6) Net field toward southwest; (7) Net field toward northeast; (8) Field removed. A limitation of this multi-pole magnetic tweezers is that its voltage driven inside is not well characterized. A joystick was used to provide qualitative control of the applied voltage, just capable to show which direction of the net total voltage is applied toward. As a result, an additional permanent magnet is needed on the top to help in obtaining repetitive response from the tethered beads. As shown in figure 6.2, there is a permanent magnet constantly set on top of the sample plane for reproducible manual manipulation around the translational stage. This is not unacceptable for DNA manipulation experiments, since it makes the optical observation of magnetic beads easier. However, to study protein conformational dynamics or enzymatic activities, a quantitative controlling of the applied magnetic field is needed, and our work is focused on making a quantitative controlling system of the four magnet poles. 6.2.2 An Improvement: Developing the New Generation Magnetic Tweezers To develop a quantitative controller of the four magnet poles, our goal is to build an electro-controlling system that can generate current to the wires on electromagnets to provide magnetic field. A simultaneous quantitative real-time digital readout is also needed. The design of quantitative voltage controller is illustrated in figure 6.4. The system includes two parts: the electric controlling module and the output module, as labeled out in the figure. Firstly, an external potential at 15V is applied through power supply. One coarse adjustment knob and one fine adjustment knob are set for different precise level of tuning potential in experiments. After going through an operational amplifier (OPA), the electric potential output in unit of Volt will be 141 translated d into curren nt output in unit u of Ampeere by the cuurrent modulle. The currrent will be tthe output to o the wires on n the electro omagnet, while 1% of thee current willl be sent to a parallel circuit to generaate a digital signal s on thee display mo odule. Figure F 6.4. A circuit diag gram of the new n controllling system ffor the quaddrupole electromagnet. This controlling c circuit c is for the purpose of quantitattively controolling one ouut of 142 the four electromagnet poles. And there are four sets of this circuit built in the controlling box independently in charge of each magnet pole. Acronyms stand for: PCB for printed circuit board; IC stands for integrated circuit; inverted triangles for ‘connected to ground’; CT for course tuning; FT for fine tuning; Polar C for polarized capacitance; Non-Polar C for non-polarized capacitance; LM324 for the operational amplifier. The newly built controller can achieve up to 1 Amperes output, which allows the electromagnet to generate more than 300 Gauss magnetic field. The controlling precision can achieve 0.01 Ampere, which can be directly shown in the digital display module on the controller. Since the capability of the electromagnet poles to manipulate paramagnetic beads has already been tested previously, this newly designed digital controller can be used to provide precise quantitative magnetic field which is beneficial for future single molecule biophysical or cell-mechanical studies using magnetic tweezers. This controller is built thanks to great helpful effort by Mr. Douglas Martin, the Design Engineer from Department of Chemistry at Bowling Green State University. And I sincerely acknowledge Mr. Jason J. Han and Mr. Alex Li from Washington State University for providing testing result of the electromagnet poles. 6.3. References 1 Strick, T.R.; Allemand, J.F.; Bensimon, D.; Bensimon, A.; Croquette, V. The Elasticity of A Single Supercoiled DNA Molecule. Science. 1996 271(5257): 1835-1837. 2 Matthews, B.D.; LaVan, D.A.; Overby, D.R.; Karavitis, J.; Ingber, D.E. Electromagnetic Needles with Submicron Pole Tip Radii for Nanomanipulation of Biomolecules and Living Cells. Appl. Phys. Lett. 2004, 85, 2968-2970. 143 3 Alenghat, F.J.; Fabry, B.; Tsai, K.Y.; Goldmann, W.H.; Ingber, D.E Analysis of Cell Mechanics in Single Vinculin-Deficient Cells Using a Magnetic Tweezer. Biochem. Biophys. Res. Commun., 2000, 277, 93-9. 4 del Rio, A.; Perez-Jimenez, R.; Liu, R.; Roca-Cusachs, P.; Fernandez, J.M.; Sheetz, M.P. Stretching Single Talin Rod Molecules Activates Vinculin Binding. Science. 2009 323 (5914): 638-641. 5 Smith, S.B.; Finzi, L.; Bustamante, C. Direct Mechanical Measurements of The Elasticity of Single DNA Molecules by Using Magnetic Beads. Science. 1992 258(5085): 1122-1126. 6 Lionnet, T.; Joubaud, S.; Lavery, R.; Bensimon, D.; Croquette, V. Wringing Out DNA. Phys. Rev. Lett. 2006, 96, 178102. 7 Bausch, A.R.; Moller, W.; Sackmann, E. Measurement of Local Viscoelasticity and Forces in Living Cells by Magnetic Tweezers. Biophys. J. 1999, 76(1) 573–579. 8 Tanase, M. Biais, N.; Sheetz, M. Magnetic Tweezers in Cell Biology. Methods Cell Biol, 2007, 83, 473–493. 9 Guilford, W.H.; Gore, R.W. A Novel Remote-sensing Isometric Force Transducer for Micromechanics Studies. Am. J. Physiol. 1992, 263, C700-7. 10 Amblard, F.; Yurke, B.; Pargellis, A.; Leibler, S. Magnetic Manipulator for Studying Local Rheology and Micromechanical Properties of Biological Systems. Rev. Sci Instrum. 1996, 67, 818. 11 Luo, C.; Glogauer, M.; ·Rossi, M.; Ferrier, J. Cell-Substrate Separation: Effect of Applied Force and Temperature. Eur. Biophys. J. 1998, 27: 9–17. 12 Haber, C.; Wirtz, D. Magnetic Tweezers for DNA Micromanipulation. Rev. Sci Instrum. 2000, 71, 4561-4570. 144 13 Gosse, C.; Croquette, V. Magnetic Tweezers: Micromanipulation and Force Measurement at the Molecular Level. Biophys. J. 2002, 82, 3314-3329. 14 Hosu, B.G.; Jakab, K.; Banki, P.; Toth, F.I.; Forgacs, G. Magnetic Tweezers for Intracellular Applications. Rev. Sci Instrum. 2003, 74, 4158. 15 Fisher, J. K.; Cummings, J.R.; Desai, K. V.; Vicci, L.; Wilde, B.; Keller, K.; Weigle, C.; Bishop, G.; Taylor, R. M.; Davis, C. W.; Boucher, R. C.; O'Brien, E. T.; Superfine, R. ThreeDimensional Force Microscope: A Nanometric Optical Tracking and Magnetic Manipulation System for the Biomedical Sciences. Rev. Sci Instrum. 2005, 76, 053711. 16 Fisher, J. K.; Cribb, J.; Desai, K. V.; Vicci, L.; Wilde, B.; Keller, K.; Taylor, R. M.; Haase, J.; Bloom, K.; O'Brien, E. T.; Superfine, R. Thin-Foil Magnetic Force System for HighNumerical-Aperture Microscopy. Rev. Sci Instrum. 2006, 77, 023702. 17 Kollmannsberger,P.; Fabry, B. High-force Magnetic Tweezers with Force Feedback for Biological Applications. Rev. Sci Instrum. 2007, 78, 114301. 18 Kanger, J.S.; Subramaniam, V.; von Driel, R. Intracellular Manipulation of Chromatin Using Magnetic Nanoparticles. Chromosom. Res. 2008, 16:511–522. 19 Bijamov, A.; Shubitidze, F.; Oliver, P.M.; Vezenov, D.V. Quantitative Modeling of Forces in Electromagnetic Tweezers. J. Appl. Phys. 2010, 108, 104701. 20 Lipfert, J.; Hao, X.; Dekker, N.R. Quantitative Modeling and Optimization of Magnetic Tweezers. Biophys J. 2009, 96(12):5040-9.