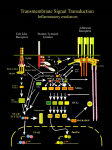
Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
This information is current as of June 18, 2017. Human Vγ2Vδ2 T Cells Augment Migration-Inhibitory Factor Secretion and Counteract the Inhibitory Effect of Glucocorticoids on IL-1 β and TNF-α Production Lisheng Wang, Hiranmoy Das, Arati Kamath, Lin Li and Jack F. Bukowski References Subscription Permissions Email Alerts This article cites 66 articles, 23 of which you can access for free at: http://www.jimmunol.org/content/168/10/4889.full#ref-list-1 Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2002 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. Downloaded from http://www.jimmunol.org/ by guest on June 18, 2017 J Immunol 2002; 168:4889-4896; ; doi: 10.4049/jimmunol.168.10.4889 http://www.jimmunol.org/content/168/10/4889 The Journal of Immunology Human V␥2V␦2 T Cells Augment Migration-Inhibitory Factor Secretion and Counteract the Inhibitory Effect of Glucocorticoids on IL-1 and TNF-␣ Production1 Lisheng Wang, Hiranmoy Das, Arati Kamath, Lin Li, and Jack F. Bukowski2 T he host response to inflammation and infection involves the production of cytokines. Three pleiotropic cytokines, macrophage migration-inhibitory factor (MIF),3 IL-1, and TNF-␣, have broad biological effects. MIF is a pituitary peptide released during the physiological stress response, a proinflammatory macrophage cytokine secreted after LPS stimulation, and a T cell product expressed as part of the Ag-dependent activation response (1, 2). MIF counteracts the inhibitory effects of glucocorticoids on TNF-␣, IL-1, IL-6, and IL-8 production by monocytes in response to stimulation with LPS in vitro (1). MIF is also involved in broad-spectrum pathophysiological reactions as an inflammatory cytokine. In mouse models, treatment with anti-MIF Ab reduces mortality of septic shock (3), suppresses endotoxininduced fatal hepatic failure (4), inhibits rheumatoid arthritis (5), and inhibits tumor growth (6, 7). MIF also augments resistance to microbial infection (8), and shows endocrine and enzymatic functions (9, 10). TNF-␣ and IL-1 are multifunctional cytokines and play pivotal roles in inflammation and infection. For example, they are considered to be master cytokines in the pathobiology of septicemia and septic shock (11, 12), and in chronic, destructive arthritis (13). Lymphocyte Biology Section, Division of Rheumatology, Immunology, and Allergy, Department of Medicine, Brigham and Women’s Hospital and Harvard Medical School, Boston, MA 02115 Received for publication September 6, 2001. Accepted for publication March 6, 2002. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact. 1 This research was supported by grants from the National Institutes of Health and the Arthritis Foundation. 2 Address correspondence and reprint requests to Dr. Jack F. Bukowski, Lymphocyte Biology Section, Division of Rheumatology, Immunology, and Allergy, Department of Medicine, Brigham and Women’s Hospital and Harvard Medical School, Smith Building, Room 526D, One Jimmy Fund Way, Boston, MA 02115. E-mail address: [email protected] 3 Abbreviations used in this paper: MIF, migration-inhibitory factor; hu-SCID, SCID mice reconstituted with human PBMC; IBA, isobutylamine; LB, Luria-Bertani; TLR, Toll-like receptor. Copyright © 2002 by The American Association of Immunologists TNF-␣ and IL-1 may contribute to pathogenesis of thyroid autoimmunity (14 –16). They may also mediate host defense responses to neuroinflammation and cell death in neurodegenerative conditions, in particular, multiple sclerosis and Parkinson’s and Alzheimer’s diseases (17–22). In addition, TNF-␣ and IL-1 have potent effects in the CNS, resulting in fever, induction of sickness behavior, and activation of the hypothalamic-pituitary-adrenal axis (reviewed in Refs. 23 and 24). Release of cytokines can be harmful and sometimes lethal to the host (25–29). Therefore, cytokine production must be reciprocally and finely tuned in vivo, and imbalance of tuning could cause immune system disorders. Glucocorticoids are one of the most potent regulators of cytokine production (30, 31). Glucocorticoids reduce the number of monocytes; lyse immature T cells; block phospholipase A2 activity; down-regulate the synthesis and secretion of IL-1, IL-6, and TNF-␣ from activated monocytes and macrophages; and inhibit cytokine-induced transcription factors, such as NF-B and AP1 (32, 33). The crucial role of glucocorticoids has been demonstrated in a number of studies in which following adrenalectomy, challenge with TNF-␣ or IL-1 at doses that would be well tolerated in adrenal-intact animals proves fatal (34). These lethal effects can be prevented by steroid treatment (34). ␥␦ T cells comprise only 2–5% of CD3⫹ cells in human peripheral blood. Approximately 70% of these ␥␦ T cells coexpress V␥2 and V␦2 TCR chains, and are uniformly reactive to nonpeptide organophosphate and alkylamine Ag, without CD1 or MHC restriction (35, 36). Several lines of evidence suggest that these cells play an important role in immunoregulation (37– 43). ␥␦ T cells prime macrophages to produce TNF-␣ in response to LPS stimulation (43), and augment production of macrophage-derived NO (44). Depletion of ␥␦ T cells results in a decrease of TNF-␣, IL-1, IFN-␥, and IL-6 gene expression in the spinal cord of mice with autoimmune encephalomyelitis (45). However, the roles of ␥␦ T cells in the immunoregulation of cytokine production are still poorly understood. It is unknown whether human ␥␦ T cells participate in regulating MIF secretion, and how ␥␦ T cells, glucocorticoids, and cytokines converge to give a unified physiological 0022-1767/02/$02.00 Downloaded from http://www.jimmunol.org/ by guest on June 18, 2017 In immune cells, proinflammatory cytokine gene expression is regulated by glucocorticoids, whereas migration-inhibitory factor (MIF), a pleiotropic cytokine, has the unique property of counteracting the inhibitory effect of glucocorticoids on TNF-␣ and IL-1 secretion. A few lines of evidence suggest that ␥␦ T cells play an important role in immunoregulation. However, it is unknown whether human ␥␦ T cells participate in regulating MIF secretion, and how ␥␦ T cells, glucocorticoids, and cytokines converge to give a unified physiological response. In this study, we demonstrate that human V␥2V␦2 T cells augment MIF secretion. Remarkably, these V␥2V␦2 T cells, functioning similarly to MIF in part, counteracted inhibition of dexamethasone on production of IL-1 and TNF-␣. SCID mice reconstituted with human PBMC that were mock depleted of V␦2 T cells and repeatedly infected with lethal dose of Escherichia coli had shorter survival time than those reconstituted with PBMC that were depleted of V␦2 T cells. Thus, human V␥2V␦2 T cells are likely to play broad-spectrum roles in immunoregulation and immunopathology by influencing MIF secretion and the immunomodulatory function of glucocorticoids. The Journal of Immunology, 2002, 168: 4889 – 4896. 4890 V␥2V␦2 T CELLS COUNTERACT INHIBITORY EFFECTS OF GLUCOCORTICOIDS response. In this study, we demonstrate that V␥2V␦2 T cells augment MIF secretion and counteract the inhibitory effect of glucocorticoids on production of IL-1 and TNF-␣, suggesting that V␥2V␦2 T cells are involved in immunoregulation. In an in vivo hu-SCID model (SCID mice reconstituted with human PBMC), human V␥2V␦2 T cells were found to participate in the pathogenesis of septic shock. Materials and Methods at the indicated time points were collected for analysis of MIF, IL-1, or TNF-␣ by ELISA. Detection of human IL-1, TNF-␣, and MIF in the tissue culture supernatant Human IL-1, TNF-␣, and MIF ELISA were performed according to the procedures recommended by the manufacturers. The detection limit of the assay was 15.6 pg/ml for IL-1, 7.8 pg/ml for TNF-␣, and 60 pg/ml for MIF. Intracellular cytokine staining mAb ascites against T cell Ags used were as follows: control mAb (P3), pan-␥␦ TCR (anti-TCR␦1), V␦1 (A13), V␦1/J␦1 (␦TCS1), V␦2 (BB3), V␥2 (7A5), and CD3 (OKT3). The specificity of these mAbs is reviewed in Porcelli et al. (46). Other reagents were purchased as follows: FITCconjugated F(ab⬘)2 goat anti-mouse IgM and IgG (catalog number AMI4708; BioSource International, Camarillo, CA); isobutylamine (IBA; catalog number I-3634; Sigma-Aldrich, St. Louis, MO); pamidronate (Novartis, East Hanover, NJ); PE-conjugated mouse anti-human TNF-␣ (catalog number 18630D; BD PharMingen, San Diego, CA), and anti-human IL-1 (catalog number IC201P; R&D Systems, Minneapolis, MN); ELISA human TNF-␣ set (catolog number 555212; BD PharMingen), and IL-1 set (catolog number 2687KI; BD PharMingen); anti-human MIF mAb (catalog number MAB289; R&D Systems); biotinylated anti-human MIF Ab (catalog number BAF289; R&D Systems); human rMIF (catalog number 289-MF; R&D Systems); LPS (catalog 201, from Escherichia coli 0111: B4; List Biological Laboratories, Campbell, CA); and Limulus amebocyte lysate (catalog number GS003; Associates of Cape Cod, Falmouth, MA). Human PBMC were cultured in RPMI medium with inclusion or exclusion of 10 nM dexamethasone in the absence of LPS (Fig. 2), or in the presence of LPS (Fig. 4). Four hours before staining with cellular surface marker, monensin (GolgiStop; BD PharMingen) that enhanced intracellular cytokine accumulation was added in the medium. Cells were washed with PBS and stained with surface marker AlexaFluor-conjugated 488 IgG control Ab, pan-TCR␦1 (Abs purified and conjugated by our laboratory), or FITCconjugated anti-CD14. After two washes, cells were fixed with 2% formaldehyde in PBS and permeabilized with 0.5% (w/v) saponin (BD PharMingen). Intracellular TNF-␣ and IL-1 were stained with PE-conjugated Ab in saponin buffer. After two washes, cells were resuspended in PBS and analyzed using FACS flow cytometer (BD Biosciences, Mountain View, CA) and Flowjo software (Tree Star, San Carlos, CA). V␦2 T cells themselves did not produce intracellular IL-1, TNF-␣, and IFN-␥ after 16 to 24 h culture in the medium containing 10% FBS or 1 g/ml LPS (47) (L. Wang, unpublished observation). PBMC Homozygous C.B-Igh-1b/Gbms-Prkdc(SCID)-Lyst(beige)N7 (SCID) male mice, 5– 6 wk old, were purchased from Taconic Farms (Germantown, NY) and maintained in microisolator cages. Animals were fed autoclaved food and water, and all manipulations were performed under laminar flow. The mice were weighed and randomly distributed into groups of 5–14 animals with equal mean body weight. E. coli (ATCC 25922) was grown in LuriaBertani (LB) broth at 37°C until the culture reached early stationary phase. E. coli was aliquoted (1 ml/vial) and stored in LB broth containing 10% glycerol at ⫺80°C until use. Before infection, E. coli were washed once with 30 ml PBS and plated on LB agar to determine CFU. The SCID mice were injected i.p. with 0.5 ml RPMI medium containing 3 ⫻ 107 human PBMC and 0.5 ml PBS containing 1–5 ⫻ 107 E. coli under aseptic conditions. For septic shock model, the mice were inoculated with E. coli twice, 1 ⫻ 107 as first dose and 5 ⫻ 107 as second dose. The interval between two bacterial infections was 20 h. The animals were observed four times per day for their survival time. Human PBMC obtained from random healthy donor leukopacks (DanaFarber Cancer Institute, Boston, MA) were isolated by Ficoll-Hypaque centrifugation (Pharmacia, Piscataway, NJ). PBMC were cryopreserved in FBS containing 10% DMSO at ⫺196°C until use. Depletion of V␥2V␦2 T cells Depletion of V␥2V␦2 T cells was performed by use of mouse anti-human V␦2 Ab (BB3), or P3, an isotype-matched mock control, and goat antimouse IgG Dynabeads M-450 (catalog number 110.06; Dynal Biotech, Oslo, Norway), according to the manufacturer’s instructions. For most depletions, P3, an isotype-matched control mAb, was substituted for the antiV␦2 mAb. For some depletions, anti-human V␦1 Ab (A13) was taken as an alternative control and substituted for the anti-V␦2 mAb. V␦2 T cells constituted 70 –90% of total ␥␦ T cells for the PBMC used in our experiments. Over 95% of V␦2 T cells, confirmed by surface marker staining and analysis of flow cytometry, were depleted. Since V␦2 T cell constituted only about 2% of CD3⫹ cells in our experiments, depletion of V␦2 T cells from PBMC with a specific V␦2 T cell Ab (BB3) did not show significant influence on percentages of CD14⫹ and other cells when analyzed by flow cytometer (data not shown). We screened several donors by two-color fluorescence and found that 100% of V␦2-bearing T cells also expressed V␥2. Although the V␦2 TCR chain paired with V␥1 or V␥3 has been described, they are extremely rare, and there is no evidence that they respond to nonpeptide Ags. Thus, the likelihood that we are studying a population other than V␥2V␦2⫹ T cells is very remote. Detection of endotoxin Endotoxin levels in the Abs and reagents were assessed with the Limulus amebocyte lysate kit according to the procedures recommended by manufacturer. The detection sensitivity of the assay was 0.03 EU/ml. Endotoxin levels in P3 (isotype-matched mock control Ab) and BB3 (V␦2 Ab) Abs were all negative at the concentration used for depletion of V␦2 T cells. Stimulation of PBMC with dead bacteria and LPS in vitro To determine whether ␥␦ T cells, under a physiological condition, play a role in regulating monocytes to produce IL-1 in presence of dexamethasone, neither LPS nor dead E. coli was applied in the experiments (Fig. 2), whereas, to determine whether ␥␦ T cells regulate PBMC to produce and secrete IL-1 and TNF-␣ in a pathological circumstance, LPS or dead E. coli was used in the experiments (Figs. 3 and 4), as described below. PBMC were washed twice after 16-h preincubation with 1 mM IBA or medium in presence or absence of dexamethasone. LPS (final concentration: 1 g/ml) or dead E. coli (inactivated at 56°C for 2 h, final concentration: 5 ⫻ 105 CFU/ml) was added to each well. The culture supernatants Engraftment and infection of SCID mice Statistics Values were expressed as means ⫾ SEM of the respective test or control group. Statistical significance between control and test groups was calculated by Student’s t test (two-tailed) and among groups by analysis of variations. Survival time was analyzed by Kaplan-Meier. Data were representative of two to four experiments. Results Human V␥2V␦2 T cells up-regulate MIF secretion MIF plays a critical role in the systemic inflammatory response by counter-regulating the inhibitory effect of circulating glucocorticoids on immune cell activation and proinflammatory cytokine production (1). To determine whether V␥2V␦2 T cells regulate MIF secretion, human PBMC were depleted or mock depleted of V␦2 T cells and subsequently stimulated with LPS. The culture supernatant was analyzed for MIF titer by ELISA. PBMC mock depleted of V␦2 T cells secreted up to 2-fold more MIF than those depleted of V␦2 T cells either in the absence (Fig. 1a) or presence (Fig. 1b) of dexamethasone, suggesting that optimal MIF secretion is dependent on presence of V␥2V␦2 T cells. Addition of 10 nM dexamethasone to the culture, in agreement with others (1, 2), inhibited MIF secretion (Fig. 1b). As an additional control, PBMC depleted of V␦1 T cells had greater levels of MIF secretion compared with V␦2-depleted PBMC, after exposure to LPS (data not shown). Optimum augmentation of MIF secretion by V␥2V␦2 T Downloaded from http://www.jimmunol.org/ by guest on June 18, 2017 Ab and Ag reagents The Journal of Immunology cells occurred at 3– 6 h after stimulation, with a major loss of titer by 9 h after stimulation (Fig. 1c). Human V␥2V␦2 T cells counteract the inhibitory effect of dexamethasone on IL-1 production Since V␥2V␦2 T cells augmented the release of MIF (Fig. 1), we speculated that these cells might also influence production and secretion of IL-1. As monocytes are a major source of IL-1, we first analyzed intracellular IL-1 production of monocytes at a physiologic concentration of dexamethasone in the absence of LPS stimulation, by use of two-color flow cytometry. In absence of dexamethasone and LPS, 41.3 and 42.6% of monocytes produced IL-1 in the absence or presence of V␥2V␦2 T cells, respectively. In contrast, when PBMC were cultivated in 10 nM dexamethasone (equivalent to a physiological level of bioactive cortisol in normal human blood), monocytes reduced their intracellular IL-1 production by up to 30-fold in the absence of V␦2 T cells (Fig. 2). This low level of intracellular IL-1 staining of monocytes in the absence of V␦2 T cells was not due to cytokine release to the extracellular medium, since there was no detectable IL-1 in the culture supernatant in the absence of LPS or dead E. coli stimulation (Fig. 3a). These data suggest that V␥2V␦2 T cells counteract the inhibitory effect of glucocorticoids on IL-1 production by monocytes under physiologic conditions, resulting in an increased storage pool of intracellular IL-1. If PBMC were stimulated with LPS for 1– 6 h, ⬎90% of CD14⫹ monocytes produced intracellular IL-1 and TNF-␣ in the absence of dexamethasone, and 30 – 60% in the presence of dexamethasone, dependent on the concentration of dexamethasone added. As a result, any potential difference in intracellular cytokine production in the presence or absence of ␥␦ T cells was veiled by this overwhelming cytokine production during short-term (1– 6 h) exposure to LPS. Since intracellular cytokine staining reflects a snapshot of accumulated cytokine by each individual cell, and only secreted cytokines affect the immunologic response, we then assessed IL-1 levels in the supernatant of PBMC in the presence of 10 nM dexamethasone. In the absence of stimulation with LPS or dead bacteria, PBMC secreted only trace amounts of IL-1, whereas, after stimulation with LPS or dead bacteria, PBMC that were mock depleted of V␦2 T cells secreted up to 2-fold more IL-1 than PBMC depleted of V␦2 T cells (Fig. 3a). If the cells were cultivated in the medium containing a natural V␥2V␦2 T cell-specific Ag, IBA (48, 49), and then stimulated with LPS, PBMC that were mock depleted of V␦2 T cells secreted up to 4-fold more IL-1 than PBMC depleted of V␦2 T cells (Fig. 3a). Replacement of LPS with dead E. coli (Fig. 3a), or replacement of IBA with the pharmaceutical V␥2V␦2 T cell-specific Ag, pamidronate (data not shown), yielded similar results. These data suggest that V␥2V␦2 T cell-specific Ags activate ␥␦ T cells, which then augment the ability of PBMC to secrete IL-1 upon exposure to LPS or dead bacteria. To determine whether MIF could reverse the inhibition of glucocorticoids on IL-1 secretion, we added 1 ng MIF to the cultures containing 10 nM dexamethasone and 1 g LPS. In this combination, PBMC depleted of V␦2 T cells resumed their IL-1 secretion nearly to the levels of PBMC that were mock depleted of V␦2 T cells (Fig. 3b). Whereas addition of exogenous MIF did not significantly augment IL-1 secretion from the PBMC that were mock depleted of V␦2 T cells, addition of 1 ng/ml MIF restored IL-1 secretion from V␦2 T cell-depleted cultures to the levels similar to those achieved in the presence of V␥2V␦2 T cells. Human V␥2V␦2 T cells counteract the inhibition by dexamethasone of TNF-␣ production and secretion Since V␥2V␦2 T cells counteracted the inhibitory effect of glucocorticoids on IL-1 production (Fig. 2), we speculated that these ␥␦ T cells might also influence production and secretion of TNF-␣. As monocytes are a major source of TNF-␣, we first analyzed the effect of 10 nM dexamethasone on the intracellular TNF-␣ production of monocytes. In the absence of V␦2 T cells, only 0.045% of CD14⫹ cells produced intracellular TNF-␣, whereas in presence Downloaded from http://www.jimmunol.org/ by guest on June 18, 2017 FIGURE 1. Human V␥2V␦2 T cells augmented MIF secretion by PBMC. Human PBMC were depleted or mock depleted of V␦2 T cells, cultivated for 16 h, and then stimulated with 1 g/ml LPS. The MIF levels in the culture supernatant were determined by ELISA. PBMC mock depleted of V␦2 T cells secreted up to 2-fold more MIF than PBMC depleted of V␦2 T cells after stimulation with LPS for 7 h (a). MIF secretion in response to LPS stimulation was inhibited by addition of 10 nM dexamethasone (b). Time course of MIF secretion in absence of dexamethasone was depicted (c). The levels of MIF before LPS stimulation were 270 –300 pg/ml in both depletion and mock depletion groups. The data of three figures were produced from different individual PBMCs. Data were representative of two experiments. 4891 4892 V␥2V␦2 T CELLS COUNTERACT INHIBITORY EFFECTS OF GLUCOCORTICOIDS of V␦2 T cells, 1.27% of CD14⫹ cells produced intracellular TNF-␣ (Fig. 2). We then determined the TNF-␣ production of monocytes in response to stimulation with LPS or heat-killed E. coli. Similar to the results obtained with IL-1 (data not shown), monocytes produced up to 2-fold more intracellular TNF-␣ in the presence than in the absence of V␥2V␦2 T cells after 18-h exposure to LPS or heat-killed E. coli (Fig. 4a). These data suggest that human V␥2V␦2 T cells, similar to mouse ␥␦ T cells (43), upregulate intracellular TNF-␣ production by human monocytes in response to stimulation with LPS or dead bacteria. We further assessed TNF-␣ secretion from PBMC in response to stimulation with LPS. Human PBMC depleted of V␦2 T cells secreted up to 2-fold less TNF-␣ as compared with PBMC that were mock depleted of V␦2 T cells, when stimulated with LPS (Fig. 4b). Since V␥2V␦2 T cells themselves did not produce and secrete TNF-␣ in response to stimulation with LPS (47), these data suggest that optimal TNF-␣ secretion by PBMC is dependent on presence of V␥2V␦2 T cells. To determine whether MIF could reverse the inhibition of glucocorticoids on TNF-␣ secretion, we added 1 ng MIF to the cultures containing 1 M dexamethasone and 1 g LPS. In this combination, PBMC depleted of V␦2 T cells resumed their TNF-␣ secretion nearly to the levels of PBMC that were mock depleted of V␦2 T cells (Fig. 4c). Whereas addition of exogenous MIF did not significantly augment TNF-␣ secretion from the PBMC that were mock depleted of V␦2 T cells, addition of 1 ng/ml MIF restored TNF-␣ secretion from V␦2 T cell-depleted cultures to levels similar to those achieved in the presence of V␥2V␦2 T cells. Taken together, the above data suggest that V␥2V␦2 T cells augment MIF secretion, counteract the inhibitory effect of glucocorticoids on production and secretion of IL-1 and TNF-␣, and influence the interaction of MIF and glucocorticoids on cytokine secretion by PBMC. V␥2V␦2 T cells play a role in septic shock in vivo Since MIF, TNF-␣, and IL-1 have been shown to be critical mediators of septic shock (3, 11), we suspected that V␥2V␦2 T cells might play a role in septic shock. To determine whether human V␥2V␦2 T cells play a role in bacterial infection in vivo, we reconstituted SCID mice with human PBMC that were either mock depleted or depleted of ␥␦ T cells, and challenged these mice with E. coli. Six SCID mice i.p. challenged with 1 ⫻ 107 E. coli all died of infection within 2 days postinfection. In contrast, five mice reconstituted with human PBMC and subsequently challenged with the same dose of E. coli all survived, suggesting that human PBMC play a crucial role against bacterial infection, and that residual mouse immune cells have negligible effects. This hu-SCID model enables us to further investigate the roles of V␥2V␦2 T cells in sepsis and septic shock. SCID mice were reconstituted with human PBMC that were depleted or mock depleted of V␦2 T cells, and subsequently infected with 1 ⫻ 107 CFU E. coli. After 20 h, the mice received a second dose of E. coli (5 ⫻ 107 CFU). In marked contrast to the experiments using low-dose bacterial infection (50), the mice receiving PBMC depleted of V␦2 T cells had a longer survival time than those receiving PBMC that were mock depleted of V␦2 T cells ( p ⫽ 0.0428, Fig. 5), suggesting that V␥2V␦2 T cells play a role in septic shock. Discussion The immunoregulatory function of V␥2V␦2 T cells is still poorly understood. In this study, we found that V␥2V␦2 T cells up-regulated MIF secretion (Fig. 1). Optimal production and secretion of IL-1 and TNF-␣ were dependent on presence of V␥2V␦2 T cells (Figs. 2, 3, and 4). Furthermore, V␥2V␦2 T cells counteracted the inhibition by glucocorticoids of IL-1 and TNF-␣ production (Figs. 2, 3b, and 4c). Recent studies suggest that physiological levels of glucocorticoids are immunomodulatory rather than solely immunosuppressive, resulting in a shift of cytokine production from a Th1- to a Th2-type pattern (30). Interruptions of this loop at any level, such as genetic, surgery, infection, or pharmacological treatments, can cause host susceptibility to infections and inflammatory diseases Downloaded from http://www.jimmunol.org/ by guest on June 18, 2017 FIGURE 2. V␥2V␦2 T cells counteracted the inhibitory effects of glucocorticoids on intracellular IL-1 and TNF-␣ production by monocytes in the absence of LPS stimulation. PBMC that were depleted or mock depleted of V␦2 T cells were cultivated in RPMI medium containing 10 nM dexamethasone (equivalent to a physiological level of bioactive cortisol in normal human blood) for 16 h in the absence of LPS stimulation. Intracellular IL-1 and TNF-␣ production by monocytes was determined by flow cytometry using a two-color staining technique. Dexamethasone inhibited the ability of monocytes to produce intracellular IL-1 and TNF-␣ by up to 30-fold in the absence of V␦2 T cells. Data were representative of three experiments. mIgG, Mouse IgG; FSC-H, forward scatter height. The Journal of Immunology (30). We speculate that overactivation of glucocorticoid regulation, as occurred in absence of V␥2V␦2 T cells, might affect severity of infectious disease. Furthermore, in absence of V␥2V␦2 T cells, MIF, TNF-␣, and IL-1, three pleiotropic and important cytokines for host resistance to bacterial infection (8, 51–57), had reduced production or secretion. The relative overactivity of glucocorticoids and reduced levels of cytokine production and secretion might account for the severity of bacterial infection in vivo. Consistent with this hypothesis, SCID mice reconstituted with human PBMC depleted of V␦2 T cells and subsequently infected FIGURE 4. V␥2V␦2 T cells augmented TNF-␣ production and secretion in response to stimulation with LPS. a, Human PBMC that were depleted or mock depleted of V␦2 T cells were cultivated in RPMI medium and stimulated with 1 g/ml LPS for 18 h. Intracellular TNF-␣ production by monocytes (stained with CD14 Ab) was determined by flow cytometry using a two-color staining technique. Following a ubiquitous burst of TNF-␣ production and secretion at the beginning of LPS stimulation, monocytes produced up to 2-fold more intracellular TNF-␣ in the presence of V␥2V␦2 T cells than in the absence of these cells 18 h after exposure to LPS. b, Human PBMC depleted of V␦2 T cells secreted up to 2-fold less TNF-␣ than PBMC that were mock depleted of V␦2 T cells when stimulated with 1 g/ml LPS for 6 h. c, PBMC were cultivated in RPMI medium containing 1 ng/ml MIF and 1 g dexamethasone for 16 h and then stimulated with 1 g/ml LPS for 6 h. MIF almost completely restored TNF-␣ secretion of V␦2 T cell-depleted cultures to the levels of PBMC cultures that were mock depleted of V␦2 T cells. Data were representative of four experiments. mIgG, Mouse IgG; FSC-H, forward scatter height. with either E. coli, Morganella morganii, or Staphylococcus aureus had much higher mortality and bacterial load than those reconstituted with human PBMC that were mock depleted of V␦2 T cells (50). V␥2V␦2 T cells remarkably counteract the inhibitory effect of glucocorticoids on IL- production, and slightly on TNF-␣ production (Fig. 2). Interestingly, the increased IL-1 in presence of V␥2V␦2 T cells was intracellularly stored in monocytes, but not extracellularly secreted in the absence of LPS or bacterial stimulation. Once exposed to LPS or dead bacteria, monocytes began to secrete IL-1 and TNF-␣, which was up-regulated by V␥2V␦2 T cells (Figs. 3 and 4). The physiological significance of this finding Downloaded from http://www.jimmunol.org/ by guest on June 18, 2017 FIGURE 3. V␥2V␦2 T cells augmented IL-1 secretion in response to stimulation with LPS. a, PBMC depleted or mock depleted of V␦2 T cells were cultivated in RPMI medium for 16 h in presence of 10 nM dexamethasone, then stimulated with either 10 ng/ml LPS or 5 ⫻ 105 heat-killed E. coli for 3 h. IL-1 levels in the culture supernatant were determined by ELISA. PBMC that were mock depleted of V␦2 T cells secreted up to 2-fold more IL-1 than PBMC depleted of V␦2 T cells. If PBMC were cultivated in RPMI medium containing a natural V␥2V␦2 T cell-specific Ag, IBA, for 16 h, and then stimulated with LPS or dead E. coli for 3 h, PBMC mock depleted of V␦2 T cells secreted up to 4-fold more IL-1 than PBMC depleted of V␦2 T cells. Replacing IBA with the pharmaceutical V␥2V␦2 T cell-specific Ag, pamidronate (data not shown), produced similar results. Stimulation with 1 g/ml LPS produced the results similar to stimulation with 10 ng/ml LPS. b, PBMC were cultivated in RPMI medium containing 10 nM dexamethasone and in the presence or absence of 1 ng/ml MIF for 16 h, and then stimulated with 1 g/ml LPS for 6 h. Addition of 1 ng/ml MIF nearly restored the IL-1 secretion from PBMC that had been depleted of V␦2 T cells, but not PBMC that had been mock depleted of V␦2 T cells. Similar results were observed in the medium containing 1 M dexamethasone. Data were representative of three experiments. 4893 4894 V␥2V␦2 T CELLS COUNTERACT INHIBITORY EFFECTS OF GLUCOCORTICOIDS FIGURE 5. V␥2V␦2 T cells play a role in septic shock. SCID mice were reconstituted with human PBMC that were depleted or mock depleted of V␦2 T cells, and subsequently infected i.p. with 1 ⫻ 107 CFU E. coli. After 20 h, the mice received a second dose of E. coli (5 ⫻ 107 CFU i.p.). The mice receiving PBMC depleted of V␦2 T cells had a longer survival time than those receiving PBMC that were mock depleted of V␦2 T cells (p ⫽ 0.0428). Data were representative of two experiments. Downloaded from http://www.jimmunol.org/ by guest on June 18, 2017 might be that, with assistance from V␥2V␦2 T cells, immune cells accumulate a high level of intracellular cytokines ready to be secreted in case of infection, making the immune response earlier and stronger. Since the prognosis of infection is dependent on the speed of immune system reaction and pathogen proliferation, early response to bacterial infection is crucial for the immune system to eliminate pathogens. Therefore, in the absence of V␥2V␦2 T cells, host resistance to bacterial infection would be impaired. On the other hand, MIF, IL-, and TNF-␣ play important roles in immunopathology. V␥2V␦2 T cells are capable of augmenting MIF secretion and counteracting the inhibitory effect of glucocorticoids on production and secretion of IL-1 and TNF-␣, thereby accounting for clinical symptoms. One piece of supportive evidence is that patients treated with the pharmaceutical V␥2V␦2 T cell-specific Ag, pamidronate, had fever and influenza-like symptoms (58). This and other aminobisphosphonate drugs have been widely used to inhibit osteoclastic bone resorption. As potent Ags, aminobisphosphonates stimulate V␥2V␦2 T cells in a TCR-dependent, MHC- and CD1-unrestricted manner (48, 49). Influenza-like symptoms occurred in these patients most likely due to the release of cytokines, since TNF-␣ and IL-1 have been demonstrated to have potent effects in the CNS, resulting in fever and sickness behavior (23, 24). These cytokines, besides being directly produced from activated V␥2V␦2 T cells, were optimally generated by other immune cells in a V␥2V␦2 T cell-dependent manner. The interactions of V␥2V␦2 T cells and glucocorticoids implicate these T cells in a broad spectrum of immunoregulatory effects. It is likely that V␥2V␦2 T cells participate in pathophysiological reactions wherever glucocorticoids are involved, such as infection, inflammatory, autoimmune, and allergic illnesses, including rheumatoid arthritis, systemic lupus erythematosus, Sjögren’s syndrome, allergic asthma, and atopic skin disease (30, 31, 59). Since activated V␥2V␦2 T cells reduce the inhibitory effect of glucocorticoids on production and secretion of TNF-␣ and IL-1, any natural V␥2V␦2 T cell-specific Ags, such as nonpeptide alkylamine (35), or organophosphate Ags (36), and drugs that activate V␥2V␦2 T cells should be considered for their interference with anti-inflammatory treatment regimens and disease outcomes. Under extreme situations, overwhelming secretion of MIF, TNF-␣, or IL-1 plays a critical role in the pathogenesis of septic shock (3, 11, 12), resulting in tissue damage, multiple organ failure, and even death. Since V␥2V␦2 T cells augment MIF secretion and counteract the inhibitory effect of glucocorticoids on production and secretion of TNF-␣ and IL-1, we suspect that these cells might play a role in septic shock. To determine whether human ␥␦ T cells mediate antibacterial effects in vivo, we developed a huSCID infective model, which has proven to be a powerful method for the study of human cells and tissues (60 – 64). We found that SCID mice inoculated with either human PBMC or Salmonella typhi all survived, whereas SCID mice receiving both PBMC and S. typhi were all dead within 10 days postinfection (L. Wang, unpublished observations), dependent on the dose of inoculated human PBMC and bacteria. These data suggest that septic shock resulted from interaction of human immune cells and bacteria. We therefore reconstituted SCID mice with human PBMC that were either mock depleted or depleted of ␥␦ T cells, then repeatedly challenged these mice with lethal doses of E. coli. The mice receiving PBMC depleted of V␦2 T cells had a longer survival time than those receiving PBMC mock depleted of V␦2 T cells (Fig. 5), suggesting that human V␥2V␦2 T cells play a role in pathogenesis of sepsis. The mechanism by which V␥2V␦2 T cells counteract inhibition of glucocorticoids on IL-1 and TNF-␣ production is unclear. Monocytes constitutively secrete MIF, which could be augmented by exposure to LPS (1, 2). T cells produce MIF as part of the Ag-dependent activation response (1, 2). In our experiments, V␦2 T cells mediating immune regulation were observed at 16 h, but not 6 h post-in vitro culture, suggesting that immune regulation of these monocytes required considerable engagement time. In addition, we had experienced technical difficulties (e.g., a very poor ratio of signal-noise) in distinguishing a small proportion of ␥␦ T cells (about 2% of CD3⫹ cells in our experiments) from whole PBMC in production of intracellular MIF. Therefore, we were unable to correlate MIF that may have been produced by a small proportion of ␥␦ T cells at a certain time point, to an inhibitory effect of glucocorticoids on IL-1 and TNF-␣ production. However, in the presence of ␥␦ T cells, MIF secretion by PBMC was augmented (Fig. 1), and the inhibitory effect of glucocorticoids on IL-1 and TNF-␣ production was counteracted (Figs. 2– 4). We speculate that the major function of V␦2 T cells is to upregulate ␣ T cells and monocytes to produce more MIF, whereas V␦2 T cell-produced MIF might constitute only a small proportion of the MIF in the supernatant. In addition, since the use of antiMIF Ab could not significantly abrogate the enhanced cytokine secretion seen in the presence of V␥2V␦2 T cells (data not shown), it is possible that, besides augmentation of MIF secretion, V␥2V␦2 T cells counteract glucocorticoid activity by other unknown pathways. This possibility is supported by the fact that addition of exogenous MIF only partially restored the deficit in IL-1 and TNF-␣ production associated with ␥␦ T cell depletion (Figs. 3b and 4c). This work demonstrates that ␥␦ T cells augment MIF secretion and counteract the inhibitory effect of glucocorticoids on IL-1 and TNF-␣ production. However, further studies are needed to determine the exact mechanisms by which this counteraction occurs. ␥␦ T cells did not produce IL-1, TNF-␣, and IFN-␥ in response to the stimulation with LPS or dead E. coli (47). However, if these ␥␦ T cells were pretreated with IBA, a V␦2 T cell-specific Ag secreted by live bacteria, then exposed to LPS, they started to produce cytokines and to expand afterward (Kamath et al., manuscript in preparation). It is possible that human ␥␦ T cells, like murine cells, express Toll-like receptors (TLRs) (65), or most likely up-regulate TLR expression on monocytes, which are involved in LPS recognition. Recently, it was found that MIF regulated monocyte immune responses through modulation of TLR4 (66). Although we were unable to detect TLR2 and TLR4 on ␥␦ T cells by flow cytometry, more sensitive methods, such as RT-PCR used for detecting TLR expression on mouse ␥␦ T cells (65), or microarray, might be alternative approaches for this purpose. The Journal of Immunology In conclusion, since V␥2V␦2 T cells augment MIF secretion and counteract inhibition of glucocorticoids on production of IL-1 and TNF-␣, we speculate that these cells play more broad-spectrum roles in immunoregulation by influencing the glucocorticoid immunomodulatory loop. References 28. Campbell, I. L. 1995. Neuropathogenic actions of cytokines assessed in transgenic mice. Int. J. Dev. Neurosci. 13:275. 29. Selin, L. K., S. M. Varga, I. C. Wong, and R. M. Welsh. 1998. Protective heterologous antiviral immunity and enhanced immunopathogenesis mediated by memory T cell populations. J. Exp. Med. 188:1705. 30. Sternberg, E. M. 2001. Neuroendocrine regulation of autoimmune/inflammatory disease. J. Endocrinol. 169:429. 31. Angeli, A., R. G. Masera, M. L. Sartori, N. Fortunati, S. Racca, A. Dovio, A. Staurenghi, and R. Frairia. 1999. Modulation by cytokines of glucocorticoid action. Ann. NY Acad. Sci. 876:210. 32. Lee, S. W., A. P. Tsou, H. Chan, J. Thomas, K. Petrie, E. M. Eugui, and A. C. Allison. 1988. Glucocorticoids selectively inhibit the transcription of the interleukin 1 gene and decrease the stability of interleukin 1 mRNA. Proc. Natl. Acad. Sci. USA 85:1204. 33. Kovalovsky, D., D. Refojo, F. Holsboer, and E. Arzt. 2000. Molecular mechanisms and Th1/Th2 pathways in corticosteroid regulation of cytokine production. J. Neuroimmunol. 109:23. 34. Bertini, R., M. Bianchi, and P. Ghezzi. 1988. Adrenalectomy sensitizes mice to the lethal effects of interleukin 1 and tumor necrosis factor. J. Exp. Med. 167: 1708. 35. Bukowski, J. F., C. T. Morita, and M. B. Brenner. 1999. Human ␥␦ T cells recognize alkylamines derived from microbes, edible plants, and tea: implications for innate immunity. Immunity 11:57. 36. Tanaka, Y., C. T. Morita, Y. Tanaka, E. Nieves, M. B. Brenner, and B. R. Bloom. 1995. Natural and synthetic non-peptide antigens recognized by human ␥␦ T cells. Nature 375:155. 37. Born, W., C. Cady, J. Jones-Carson, A. Mukasa, M. Lahn, and R. O’Brien. 1999. Immunoregulatory functions of ␥␦ T cells. Adv. Immunol. 71:77. 38. Peterman, G. M., C. Spencer, A. I. Sperling, and J. A. Bluestone. 1993. Role of ␥␦ T cells in murine collagen-induced arthritis. J. Immunol. 151:6546. 39. Harrison, L. C., M. Dempsey-Collier, D. R. Kramer, and K. Takahashi. 1996. Aerosol insulin induces regulatory CD8 ␥␦ T cells that prevent murine insulindependent diabetes. J. Exp. Med. 184:2167. 40. Ke, Y., K. Pearce, J. P. Lake, H. K. Ziegler, and J. A. Kapp. 1997. ␥␦ T lymphocytes regulate the induction and maintenance of oral tolerance. J. Immunol. 158:3610. 41. McMenamin, C., C. Pimm, M. McKersey, and P. G. Holt. 1994. Regulation of IgE responses to inhaled antigen in mice by antigen-specific ␥␦ T cells. Science 265:1869. 42. Mengel, J., F. Cardillo, L. S. Aroeira, O. Williams, M. Russo, and N. M. Vaz. 1995. Anti-␥␦ T cell Ab blocks the induction and maintenance of oral tolerance to ovalbumin in mice. Immunol. Lett. 48:97. 43. Nishimura, H., M. Emoto, K. Hiromatsu, S. Yamamoto, K. Matsuura, H. Gomi, T. Ikeda, S. Itohara, and Y. Yoshikai. 1995. The role of ␥␦ T cells in priming macrophages to produce tumor necrosis factor-␣. Eur. J. Immunol. 25:1465. 44. Jones-Carson, J., A. Vazquez-Torres, H. C. van der Heyde, T. Warner, R. D. Wagner, and E. Balish. 1995. ␥␦ T cell-induced nitric oxide production enhances resistance to mucosal candidiasis. Nat. Med. 1:552. 45. Rajan, A. J., J. D. Klein, and C. F. Brosnan. 1998. The effect of ␥␦ T cell depletion on cytokine gene expression in experimental allergic encephalomyelitis. J. Immunol. 160:5955. 46. Porcelli, S., M. B. Brenner, and H. Band. 1991. Biology of the human ␥␦ T-cell receptor. Immunol. Rev. 120:137. 47. Wang, L., H. Das, A. Kamath, and J. Bukowski. 2001. Human V␥2V␦2 T cells produce IFN-␥ and TNF-␣ with an on/off/on cycling pattern in response to live bacterial products. J. Immunol. 167:6195. 48. Das, H., L. Wang, A. Kamath, and J. Bukowski. 2001. V␥2 and V␦2 T cell receptor-mediated recognition of aminobisphosphonates. Blood 98:1616. 49. Miyagawa, F., Y. Tanaka, S. Yamashita, and N. Minato. 2001. Essential requirement of antigen presentation by monocyte lineage cells for the activation of primary human ␥␦ T cells by aminobisphosphonate antigen. J. Immunol. 166: 5508. 50. Wang, L., A. Kamath, H. Das, L. Li, and J. Bukowski. 2001. Antibacterial effect of human V␥2V␦2 T cells in vivo. J. Clin. Invest. 108:1349. 51. Vazquez-Torres, A., G. Fantuzzi, C. K. Edwards III, C. A. Dinarello, and F. C. Fang. 2001. Defective localization of the NADPH phagocyte oxidase to Salmonella-containing phagosomes in tumor necrosis factor p55 receptor-deficient macrophages. Proc. Natl. Acad. Sci. USA 98:2561. 52. Zhao, Y. X., H. Zhang, B. Chiu, U. Payne, and R. D. Inman. 1999. Tumor necrosis factor receptor p55 controls the severity of arthritis in experimental Yersinia enterocolitica infection. Arthritis Rheum. 42:1662. 53. Netea, M. G., L. J. van Tits, J. H. Curfs, F. Amiot, J. F. Meis, J. W. van der Meer, and B. J. Kullberg. 1999. Increased susceptibility of TNF-␣ lymphotoxin-␣ double knockout mice to systemic candidiasis through impaired recruitment of neutrophils and phagocytosis of Candida albicans. J. Immunol. 163:1498. 54. Simms, H. H., and R. D’Amico. 1997. Studies on polymorphonuclear leukocyte bactericidal function: the role of exogenous cytokines. Shock 7:84. 55. Evans, T. J., L. D. Buttery, A. Carpenter, D. R. Springall, J. M. Polak, and J. Cohen. 1996. Cytokine-treated human neutrophils contain inducible nitric oxide synthase that produces nitration of ingested bacteria. Proc. Natl. Acad. Sci. USA 93:9553. 56. Mancilla, J., P. Garcia, and C. A. Dinarello. 1993. The interleukin-1 receptor Downloaded from http://www.jimmunol.org/ by guest on June 18, 2017 1. Calandra, T., and R. Bucala. 1997. Macrophage migration inhibitory factor (MIF): a glucocorticoid counter-regulator within the immune system. Crit. Rev. Immunol. 17:77. 2. Calandra, T., L. A. Spiegel, C. N. Metz, and R. Bucala. 1998. Macrophage migration inhibitory factor is a critical mediator of the activation of immune cells by exotoxins of Gram-positive bacteria. Proc. Natl. Acad. Sci. USA 95:11383. 3. Calandra, T., B. Echtenacher, D. L. Roy, J. Pugin, C. N. Metz, L. Hultner, D. Heumann, D. Mannel, R. Bucala, and M. P. Glauser. 2000. Protection from septic shock by neutralization of macrophage migration inhibitory factor. Nat. Med. 6:164. 4. Kobayashi, S., J. Nishihira, S. Watanabe, and S. Todo. 1999. Prevention of lethal acute hepatic failure by antimacrophage migration inhibitory factor Ab in mice treated with bacille Calmette-Guerin and lipopolysaccharide. Hepatology 29:1752. 5. Leech, M., C. Metz, P. Hall, P. Hutchinson, K. Gianis, M. Smith, H. Weedon, S. R. Holdsworth, R. Bucala, and E. F. Morand. 1999. Macrophage migration inhibitory factor in rheumatoid arthritis: evidence of proinflammatory function and regulation by glucocorticoids. Arthritis Rheum. 42:1601. 6. Abe, R., T. Peng, J. Sailors, R. Bucala, and C. N. Metz. 2001. Regulation of the CTL response by macrophage migration inhibitory factor. J. Immunol. 166:747. 7. Mitchell, R. A., and R. Bucala. 2000. Tumor growth-promoting properties of macrophage migration inhibitory factor (MIF). Semin. Cancer Biol. 10:359. 8. Juttner, S., J. Bernhagen, C. N. Metz, M. Rollinghoff, R. Bucala, and A. Gessner. 1998. Migration inhibitory factor induces killing of Leishmania major by macrophages: dependence on reactive nitrogen intermediates and endogenous TNF-␣. J. Immunol. 161:2383. 9. Kleemann, R., A. Hausser, G. Geiger, R. Mischke, A. Burger-Kentischer, O. Flieger, F. J. Johannes, T. Roger, T. Calandra, A. Kapurniotu, et al. 2000. Intracellular action of the cytokine MIF to modulate AP-1 activity and the cell cycle through Jab1. Nature 408:211. 10. Waeber, G., T. Calandra, C. Bonny, and R. Bucala. 1999. A role for the endocrine and pro-inflammatory mediator MIF in the control of insulin secretion during stress. Diabetes Metab. Res. Rev. 15:47. 11. Glauser, M. P., G. Zanetti, J. D. Baumgartner, and J. Cohen. 1991. Septic shock: pathogenesis. Lancet 338:732. 12. Das, U. N. 2000. Critical advances in septicemia and septic shock. Crit. Care 4:290. 13. Arend, W. P., and C. Gabay. 2000. Physiologic role of interleukin-1 receptor antagonist. Arthritis Res. 2:245. 14. Drugarin, D., S. Negru, A. Koreck, I. Zosin, and C. Cristea. 2000. The pattern of a T(H)1 cytokine in autoimmune thyroiditis. Immunol. Lett. 71:73. 15. Smith, T. J., D. Sciaky, R. P. Phipps, and T. A. Jennings. 1999. CD40 expression in human thyroid tissue: evidence for involvement of multiple cell types in autoimmune and neoplastic diseases. Thyroid 9:749. 16. Rasmussen, A. K., K. Bendtzen, and U. Feldt-Rasmussen. 2000. Thyrocyte-interleukin-1 interactions. Exp. Clin. Endocrinol. Diabetes 108:67. 17. Kassiotis, G., and G. Kollias. 2001. Uncoupling the proinflammatory from the immunosuppressive properties of tumor necrosis factor (TNF) at the p55 TNF receptor level: implications for pathogenesis and therapy of autoimmune demyelination. J. Exp. Med. 193:427. 18. Tejada-Simon, M. V., J. Hong, V. M. Rivera, and J. Z. Zhang. 2001. Reactivity pattern and cytokine profile of T cells primed by myelin peptides in multiple sclerosis and healthy individuals. Eur. J. Immunol. 31:907. 19. Grunblatt, E., S. Mandel, and M. B. Youdim. 2000. Neuroprotective strategies in Parkinson’s disease using the models of 6-hydroxydopamine and MPTP. Ann. NY Acad. Sci. 899:262. 20. Rhodin, J., T. Thomas, M. Bryant, and E. T. Sutton. 2000. Animal model of Alzheimer-like vascular pathology and inflammatory reaction. Ann. NY Acad. Sci. 903:345. 21. Combs, C. K., D. E. Johnson, J. C. Karlo, S. B. Cannady, and G. E. Landreth. 2000. Inflammatory mechanisms in Alzheimer’s disease: inhibition of -amyloid-stimulated proinflammatory responses and neurotoxicity by PPAR␥ agonists. J. Neurosci. 20:558. 22. Rothwell, N. J., and G. N. Luheshi. 2000. Interleukin 1 in the brain: biology, pathology and therapeutic target. Trends Neurosci. 23:618. 23. Rothwell, N. J., and S. J. Hopkins. 1995. Cytokines and the nervous system. II. Actions and mechanisms of action. Trends Neurosci. 18:130. 24. Hopkins, S. J., and N. J. Rothwell. 1995. Cytokines and the nervous system. I. Expression and recognition. Trends Neurosci. 18:83. 25. Wysocka, M., M. Kubin, L. Q. Vieira, L. Ozmen, G. Garotta, P. Scott, and G. Trinchieri. 1995. Interleukin-12 is required for interferon-␥ production and lethality in lipopolysaccharide-induced shock in mice. Eur. J. Immunol. 25:672. 26. Vassalli, P. 1992. The pathophysiology of tumor necrosis factors. Annu. Rev. Immunol. 10:411. 27. Hayden, F. G., R. Fritz, M. C. Lobo, W. Alvord, W. Strober, and S. E. Straus. 1998. Local and systemic cytokine responses during experimental human influenza A virus infection: relation to symptom formation and host defense. J. Clin. Invest. 101:643. 4895 4896 57. 58. 59. 60. 61. V␥2V␦2 T CELLS COUNTERACT INHIBITORY EFFECTS OF GLUCOCORTICOIDS antagonist can either reduce or enhance the lethality of Klebsiella pneumoniae sepsis in newborn rats. Infect. Immun. 61:926. Shi, J., R. D. Goodband, M. M. Chengappa, J. L. Nelssen, M. D. Tokach, D. S. McVey, and F. Blecha. 1994. Influence of interleukin-1 on neutrophil function and resistance to Streptococcus suis in neonatal pigs. J. Leukocyte Biol. 56:88. Kunzmann, V., E. Bauer, and M. Wilhelm. 1999. ␥/␦ T-cell stimulation by pamidronate. N. Engl. J. Med. 340:737. Lahn, M., A. Kanehiro, K. Takeda, A. Joetham, J. Schwarze, G. Kohler, R. O’Brien, E. W. Gelfand, W. Born, and A. Kanehio. 1999. Negative regulation of airway responsiveness that is dependent on ␥␦ T cells and independent of ␣ T cells. Nat. Med. 5:1150. Murphy, W. J., D. D. Taub, and D. L. Longo. 1996. The huPBL-SCID mouse as a means to examine human immune function in vivo. Semin. Immunol. 8:233. McCune, J. M. 1996. Development and applications of the SCID-hu mouse model. Semin. Immunol. 8:187. 62. Feuerer, M., P. Beckhove, L. Bai, E. F. Solomayer, G. Bastert, I. J. Diel, C. Pedain, M. Oberniedermayr, V. Schirrmacher, and V. Umansky. 2001. Therapy of human tumors in NOD/SCID mice with patient-derived reactivated memory T cells from bone marrow. Nat. Med. 7:452. 63. Stoddart, C. A., T. J. Liegler, F. Mammano, V. D. Linquist-Stepps, M. S. Hayden, S. G. Deeks, R. M. Grant, F. Clavel, and J. M. McCune. 2001. Impaired replication of protease inhibitor-resistant HIV-1 in human thymus. Nat. Med. 7:712. 64. Ohashi, K., P. L. Marion, H. Nakai, L. Meuse, J. M. Cullen, B. B. Bordier, R. Schwall, H. B. Greenberg, J. S. Glenn, and M. A. Kay. 2000. Sustained survival of human hepatocytes in mice: a model for in vivo infection with human hepatitis B and hepatitis ␦ viruses. Nat. Med. 6:327. 65. Mokuno, Y., T. Matsuguchi, M. Takano, H. Nishimura, J. Washizu, T. Ogawa, O. Takeuchi, S. Akira, Y. Nimura, and Y. Yoshikai. 2000. Expression of Toll-like receptor 2 on ␥␦ T cells bearing invariant V␥6/V␦1 induced by Escherichia coli infection in mice. J. Immunol. 165:931. 66. Roger, T., J. David, M. P. Glauser, and T. Calandra. 2001. MIF regulates innate immune responses through modulation of Toll-like receptor 4. Nature 414:920. Downloaded from http://www.jimmunol.org/ by guest on June 18, 2017