Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Neuropsychopharmacology wikipedia , lookup
Compounding wikipedia , lookup
Pharmacognosy wikipedia , lookup
Clinical trial wikipedia , lookup
Neuropharmacology wikipedia , lookup
Drug interaction wikipedia , lookup
Prescription drug prices in the United States wikipedia , lookup
Drug design wikipedia , lookup
Pharmacogenomics wikipedia , lookup
Prescription costs wikipedia , lookup
Pharmaceutical industry wikipedia , lookup
Drug discovery wikipedia , lookup
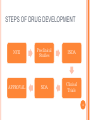
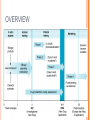
CLINICAL RESEARCH CONTENTS Drug Development Process Pre – Clinical Studies Clinical Trials Phase I Phase II Phase III Phase IV 2 DRUG DEVELOPMENT Drug development is a term used to define the entire process of bringing a new drug or device to the market. It includes: drug discovery product development pre-clinical research (microorganisms/animals) clinical trials (on humans) 3 STEPS OF DRUG DEVELOPMENT NCE Preclinical Studies INDA APPROVAL NDA Clinical Trials 4 NEW CHEMICAL ENTITIES (NCE’s) New Chemical Entities are compounds which emerge from the process of drug discovery. Little will be known about the safety, toxicity, pharmacokinetics and metabolism of this NCE in humans. It becomes necessary to assess all of these parameters before conducting human clinical trials. Pre- Clinical studies are performed 5 INVESTIGATIONAL NEW DRUG APPLICATION (INDA) Application filed with the FDA after completion of pre-clinical and prior to human testing. The IND application contains: Preclinical data: Animal Pharmacology and Toxicology Studies Chemistry and Manufacturing Information 6 INVESTIGATIONAL NEW DRUG APPLICATION (INDA) Clinical Trial Protocols & Investigator Information: Detailed protocols for proposed clinical studies to assess whether the initial-phase trials will expose subjects to unnecessary risks. Unless the FDA specifically objects, the IND is automatically allowed after 30 days and clinical trials can begin. 7 NEW DRUG APPLICATION (NDA) Submitted after completion of Phase I, Phase II, Phase III studies of Clinical Trials Application filed with the FDA for sale and marketing of the drug product. 8 NEW DRUG APPLICATION (NDA) Following information is reviewed through NDA: 1. Is the drug safe and effective in its proposed use(s) when used as directed, and do the benefits of the drug outweigh the risks? 2. Is the drug’s proposed labeling (package insert) appropriate, and what should it contain? 3. Are the methods used in manufacturing (GMP) the drug and the controls used to maintain the drug’s quality adequate to preserve the drug’s identity, strength, quality, and purity? 9 OVERVIEW 10 PRE- CLINICAL STUDIES Drugs that pass the initial screening and profiling procedures are carefully evaluated for potential risks before and during clinical testing. Pre-clinical studies include Safety and Toxicity testing. Generally conducted on animals, microorganisms or tissue cultures. 11 PRE- CLINICAL STUDIES Toxicity studies: acute toxicity - effects of large single doses up to the lethal level. Usually two species, two routes, single dose. subacute and chronic toxicity- effects of multiple doses, especially for drug that is intended for prolonged use in humans effects on reproductive functions, including teratogenicity and postnatal development Carcinogenicity: Two years, two species. Mutagenicity, Investigative toxicology 12 PRE- CLINICAL STUDIES In addition to toxicity studies: "no-effect" dose—the maximum dose at which a specified toxic effect is not seen minimum lethal dose—the smallest dose that is observed to kill any animal LD50 – Lethal Dose at 50% ED50 – Effective Dose at 50% 13 PRE- CLINICAL STUDIES ED LD 100 % subjects 50 0 0 • Relatively safe DRUG DOSE X 14 PRE-CLINICAL STUDIES ED LD 100 % subjects 50 0 0 • Less safe drug DRUG DOSE X 15 LIMITATIONS • • • • • Toxicity testing is time-consuming and expensive. Large numbers of animals are needed to obtain valid preclinical data. Cell and tissue culture in vitro methods are increasingly being used, but their predictive value is still severely limited. Extrapolation of toxicity data from animals to humans is not completely reliable. For statistical reasons, rare adverse effects are unlikely to be detected. 16 CLINICAL TRIALS • • Clinical trials are conducted on human volunteers to allow safety and efficacy data to be collected for health interventions The most commonly performed clinical trials evaluate new drugs, medical devices, biologics, psychological therapies or other interventions. 17 WHY ARE CLINICAL STUDIES PERFORMED? No chemical can be certified as completely "safe". Every chemical is toxic at some dosage It is important to estimate the risk associated with exposure to the chemical under specified conditions by performing appropriate tests. Clinical trials may be required before the national regulatory authority approves marketing of the drug or device, or a new dose of the drug, for use on patients. 18 WHEN IS CLINICAL TRIAL NECESSARY? 1. 2. 3. 4. Assess safety and efficacy of New Chemical Entity Assess safety and effectiveness of a different dose (e.g., 10 mg dose instead of 5 mg dose) Assess safety and efficacy of an already marketed medication or device for a new indication i.e. a disease for which the drug is not specifically approved To compare the effectiveness in patients with a specific disease of two or more already approved or common interventions for that disease 19 CLINICAL TRIALS I. II. III. IV. Phase I – Healthy Volunteers Phase II – Target Patient Volunteers Phase III – Larger Number of Patients Phase IV – After marketing 20 PHASE I • • • In phase I, the effects of the drug is checked in a small number of healthy volunteers. If the drug is expected to have significant toxicity, as is often the case in cancer and AIDS therapy, volunteer patients with the disease are used in phase I rather than normal volunteers. Phase I trials are done to determine whether humans and animals show significantly different responses to the drug and to establish safe clinical dosage range. 21 PHASE I • • • These trials are non-blind or "open," i.e, both the investigators and the subjects know what is being given. Many predictable toxicities are detected in this phase. This phase includes trials designed to assess the safety (pharmacovigilance), tolerability, pharmacokinetics, and pharmacodynamics of a drug. 22 PHASE I These trials are often conducted in an inpatient clinic, where the subject can be observed by full-time staff. The subject who receives the drug is usually observed until several half-lives of the drug have passed. Phase I trials also include dose-ranging, also called dose escalation, studies so that the appropriate dose for therapeutic use can be found. 23 PHASE I SAD : Single Ascending Dose small groups of subjects are given a single dose of the drug while they are observed and tested for a period of time. If they do not exhibit any adverse side effects, and the pharmacokinetic data is roughly in line with predicted safe values, the dose is escalated, and a new group of subjects is then given a higher dose. 24 PHASE I SAD : Single Ascending Dose This is continued until pre-calculated pharmacokinetic safety levels are reached, or intolerable side effects start showing up At this point the drug is said to have reached the Maximum tolerated dose (MTD). 25 PHASE I MAD: Multiple Ascending Dose studies In these studies, a group of patients receives multiple low doses of the drug Samples of blood and other fluids are collected at various time points and analyzed to understand how the drug is processed within the body. The dose is subsequently escalated for further groups, up to a predetermined level. 26 PHASE I Food effect A short trial designed to investigate any differences in absorption of the drug by the body, caused by eating before the drug is given. These studies are usually run as a crossover study, with volunteers being given two identical doses of the drug on different occasions; one while fasted, and one after being fed. 27 PHASE II • • • In phase II, the drug is studied for the first time in patients with the target disease to determine its efficacy. A small number of patients (100–200) are studied in great detail. When the development process for a new drug fails, this usually occurs during Phase II trials when the drug is discovered not to work as planned, or to have toxic effects. 28 PHASE II Phase II studies are sometimes divided into Phase IIA and Phase IIB. Phase IIA: designed to assess dosing requirements (how much drug should be given). Phase IIB: designed to study efficacy (how well the drug works at the prescribed dose(s)). 29 PHASE II A single-blind design is often used, with an inert placebo medication and an older active drug (positive control) in addition to the investigational agent. Phase II trials are usually done in special clinical centers (ex: university hospitals). A broader range of toxicities may be detected in this phase. 30 PHASE III • • • In phase III, the drug is evaluated in much larger numbers of patients—sometimes thousands—to further establish safety and efficacy. Using information gathered in phases I and II, phase III trials are designed to minimize errors caused by placebo effects, variable course of the disease, etc. Therefore, double-blind and crossover techniques are frequently used. 31 PHASE III • • • Phase III studies can be difficult to design and execute and are usually expensive because of the large numbers of patients involved and the masses of data that must be collected and analyzed. The investigators are usually specialists in the disease being treated. Certain toxic effects—especially those caused by immunologic processes—may first become apparent in phase III. 32 PHASE IV • • 1. 2. 3. Phase IV trial is also known as Post Marketing Surveillance Trial. Phase IV studies may be required by regulatory authorities or may be undertaken by the sponsoring company for competitive (finding a new market for the drug) To check for drug interactions with other drugs certain population groups such as pregnant women, who are unlikely to subject 33 themselves to trials PHASE IV • • • This constitutes monitoring the safety of the new drug under actual conditions of use in large numbers of patients. The sample size required to disclose druginduced events or toxicities is very large for such rare events. For example, several hundred thousand patients may have to be exposed before the first case is observed of a toxicity that occurs with an average incidence of 1 in 10,000. 34 PHASE IV • • • • Careful and complete reporting of toxicity by physicians after marketing begins is very important. Low incidence drug effects will not generally be detected before phase 4 no matter how carefully the studies are executed. Phase 4 has no fixed duration. Adverse effects detected by Phase IV trials may result in withdrawal or restriction of a drug. 35 Comparison Phase I Phase II Phase III Phase IV Healthy Volunteers Target disease Patients Target disease Patients Mixed Sample Size 20 – 80 200 – 300 100 – 1000 1000 < Duration 1 month Several Months Several Years Ongoing Design Non blind Single blind Double blind Population Uncontrolled 36 37