Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Coronary artery disease wikipedia , lookup
Heart failure wikipedia , lookup
Hypertrophic cardiomyopathy wikipedia , lookup
Quantium Medical Cardiac Output wikipedia , lookup
Cardiac surgery wikipedia , lookup
Myocardial infarction wikipedia , lookup
Cardiac contractility modulation wikipedia , lookup
Electrocardiography wikipedia , lookup
Ventricular fibrillation wikipedia , lookup
Heart arrhythmia wikipedia , lookup
Arrhythmogenic right ventricular dysplasia wikipedia , lookup
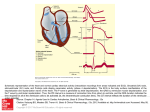
Larger Cell Size in Rabbits With Heart Failure Increases Myocardial Conduction Velocity and QRS Duration Rob F. Wiegerinck, MSc; Arie O. Verkerk, PhD; Charly N. Belterman, RA; Toon A.B. van Veen, PhD; Antonius Baartscheer, PhD; Tobias Opthof, PhD; Ronald Wilders, PhD; Jacques M.T. de Bakker, PhD; Ruben Coronel, MD, PhD Downloaded from http://circ.ahajournals.org/ by guest on June 18, 2017 Background—Patients with heart failure (HF) have an increased QRS duration, usually attributed to decreased conduction velocity (CV) due to ionic remodeling but which may alternatively result from increased heart size or cellular uncoupling. We investigated the relationship between QRS width, heart size, intercellular coupling, and CV in a rabbit model of moderate HF and in computer simulations. Methods and Results—HF was induced by pressure-volume overload. Heart weight (21.1⫾0.5 versus 10.2⫾0.4 g, mean⫾SEM; P⬍0.01) and QRS duration (58⫾1 versus 50⫾1 ms; P⬍0.01) were increased in HF versus control. Longitudinal CV (L; 79⫾2 versus 67⫾4 cm/s; P⬍0.01) and transversal subepicardial CV (T; 43⫾2 versus 37⫾2 cm/s; P⬍0.05) were higher in HF than in controls. Transmural CV (TM) was unchanged (25⫾2 versus 24⫾1 cm/s; P⫽NS). Patch-clamp experiments demonstrated that sodium current was unchanged in HF versus control. Immunohistochemical experiments revealed that connexin43 content was reduced in midmyocardium but unchanged in subepicardium. Myocyte dimensions were increased in HF by ⬇30%. Simulated strands of mammalian ventricular cells (Luo-Rudy dynamic model) revealed increased L and T with increased myocyte size; however, increased CV could not compensate for increased strand size of longitudinally coupled cells, and consequently, total activation time was longer. Conclusions—Increased myocyte size combined with the observed expression pattern of connexin43 yields increased L and T and unchanged TM in our nonischemic model of HF. A hypertrophied left ventricle together with insufficiently increased L and unaltered TM results in a prolonged QRS duration. (Circulation. 2006;113:806-813.) Key Words: hypertrophy 䡲 heart failure 䡲 electrocardiography 䡲 myocytes 䡲 conduction P atients with heart failure (HF) have a poor prognosis. Mortality is high, and ⬇50% of patients with HF die suddenly, which suggests an arrhythmogenic origin.1,2 Patients with HF often reveal increased QRS duration3,4 and those with QRS duration ⬎150 ms have a higher cardiac mortality rate than patients with shorter QRS duration.4,5 Increased QRS duration is commonly interpreted as slowing of ventricular conduction. Although slow conduction is one of the prerequisites for the occurrence of reentrant arrhythmias6 and therefore may explain the increased propensity of patients with HF for lifethreatening arrhythmias,1,2 studies on conduction velocity (CV) in HF are contradictory. at day 150 after aortic banding; however, in the same model, CV was increased after 50 days of aortic banding.11 This underscores the dynamic nature of HF. In 2 other studies that included patients with HF, an increased CV was measured12,13 as well. Increased CV was also found in hypertrophied hearts.11–13 In hypertrophy, cell size increases. We hypothesized that increased cell size underlies both the increase in CV and the increase in QRS duration. The latter would increase because it takes more time to activate the hypertrophied ventricles, despite the increase in CV. The aim of the present study was to document CV changes and to correlate CV with QRS duration, connexin43 (Cx43) content, and cell size in an established nonischemic model of moderate HF,14 in which hypertrophy is the predominant feature and in which electrical remodeling is negligible. The functional effects of the experimentally observed changes in cell size were assessed with computer simulations at various preset values of intercellular coupling. Clinical Perspective p 813 In animal studies, unchanged CV7,8 and decreased CV9 have been reported in the setting of HF. QRS duration was increased10 and CV decreased11 in a guinea pig model of HF Received May 31, 2005; revision received December 7, 2005; accepted December 8, 2005. From the Departments of Experimental Cardiology (R.F.W., A.O.V., C.N.B., A.B., T.O., J.M.T.d.B., R.C.) and Physiology (A.O.V., R.W.), Academic Medical Center, University of Amsterdam, Amsterdam, the Netherlands; the Department of Medical Physiology (T.A.B.v.V., T.O., J.M.T.d.B.), University Medical Center Utrecht, Utrecht, the Netherlands; and the Interuniversity Cardiology Institute of the Netherlands (J.M.T.d.B.), Utrecht, the Netherlands. Correspondence to Rob F. Wiegerinck, MSc, Department of Experimental Cardiology, Molecular & Experimental Cardiology Group, Academic Medical Center, Room M0-109, Meibergdreef 9, 1105 AZ Amsterdam, The Netherlands. E-mail [email protected] © 2006 American Heart Association, Inc. Circulation is available at http://www.circulationaha.org DOI: 10.1161/CIRCULATIONAHA.105.565804 806 Wiegerinck et al Methods Rabbit Model of HF Downloaded from http://circ.ahajournals.org/ by guest on June 18, 2017 The experimental protocol complied with the “Guide for the Care and use of Laboratory Animals” published by the US National Institutes of Health (NIH publication 85-23, revised 1985) and was approved by the institutional animal experiments committee. HF was induced by combined volume and pressure overload in 2 sequential surgical procedures in New Zealand White rabbits as described previously.14 In that study,14 differences between nonoperated and sham-operated animals were negligible. Thus, for the present study, control rabbits were not sham-operated. Before the first operation, a 3-lead ECG was recorded in rabbits premedicated with ketamine (100 mg/mL, 0.7 mL/kg body weight IM) and xylazine (20 mg/mL, 0.3 mL/kg body weight IM). Control and HF rabbits (at 8 months of age or when HF with overt signs of dyspnea developed) were weighed, a 3-lead ECG was recorded, and left ventricular end-diastolic pressure was measured as described previously.14 Rabbits were heparinized (1000 IU IV) and killed by injection of pentobarbital (60 mg/mL, 5 mL, IV). Hearts were isolated and submerged in Tyrode’s solution (4°C; for composition, see below). Presence of ascites and heart and lung weights were documented. Conduction, QRS Width, and Heart Failure 807 was achieved by using low-resistance pipettes (1.0 to 2.0 M⍀) and Rs and Cm compensation ⬎80%. In the cell-attached macropatch experiments, the pipette and bath solutions contained (in mmol/L) NaCl 140, KCl 5.4, CaCl2 1.8, MgCl2 1.0, glucose 5.5, and HEPES 5.0 (pH 7.3 [NaOH]). The pipette command potential was corrected for the resting membrane potential (Vm) of the myocytes, which was ⫺81.5⫾0.9 and ⫺81.7⫾0.6 mV in control (n⫽37) and failing (n⫽32) myocytes, respectively, as determined in a separate set of whole-cell patchclamp experiments. INa was defined as the difference between peak current and the current at the end of the depolarizing voltage step. Myocyte Dimensions The size of individual left ventricular myocytes was determined in 6 control and 7 HF animals. Enzymatically isolated myocytes were studied in a cell chamber on the stage of an inverted microscope during superfusion with a HEPES solution containing (in mmol/L) NaCl 148, NaHCO3 1.0, KHCO3 3.3, KH2PO4 1.4, MgCl2 1.9, CaCl2 1.3, glucose 11.0, HEPES 16.8 (37°C, pH 7.3 [NaOH]). Cellular length and width were measured at a resolution of 3 m in 50 randomly selected viable rod-shaped myocytes per heart. Immunohistochemistry and Histology Perfusion of Hearts After excision of the heart, the aorta was prepared free from adjacent tissue. In control rabbits, a cannula was inserted into the aorta to allow retrograde perfusion of the heart. Because retrograde perfusion is impossible owing to aortic insufficiency in HF, both coronary arteries were cannulated separately. The hearts were perfused with Tyrode’s solution containing (in mmol/L) 130 NaCl, 5.6 KCl, 2.2 CaCl2, 0.55 MgCl2, 24.2 NaHCO3, and 11.1 glucose, saturated with 95% O2 and 5% CO2. Myocardial temperature was maintained at 37°C. Activation Mapping and CV Activation times (ATs) were derived from local electrograms recorded with an epicardial multielectrode (247 unipolar electrode terminals, 1.5⫻1 mm interelectrode distance) and/or with 4 intramural needles (8 electrode terminals/needle, interelectrode distance 0.3 mm) inserted 1 cm apart in a square on the left ventricular free wall. Electrodes were connected to a data-acquisition system (Biosemi Mark-6). A virtual ground electrode was connected to the aortic root. Data of 1.8-second episodes (sampling interval 0.5 ms) were stored on disk. Local AT, defined as the moment of the maximum negative dV/dt, was determined with a custom interactive analysis program.15 Pacing from the center of the epicardial multielectrode or from the most distal located electrode on a needle was performed with rectangular current pulses at diastolic stimulation threshold (2-ms duration) at a basic cycle length of 250 ms. Subepicardial activation maps were constructed, and longitudinal (L) and transversal (T) CVs were calculated from the ellipsoid activation pattern as described previously.16 Transmural CV (TM, perpendicular to the epicardial surface) was calculated from local ATs measured on 1 needle. Sodium Current The fast sodium current (INa) is responsible for the action potential upstroke and is important for CV.17 Therefore, INa was evaluated by whole-cell patch-clamp (20°C) and cell-attached macropatch recordings (37°C).18 Myocytes were enzymatically isolated as described previously.19 Currents were filtered at 5 kHz and digitized at 10 kHz. Voltage control, data acquisition, and analysis were accomplished with custom software. The INa current-voltage relationship was determined by the voltage-clamp protocols diagrammed in Figure 2. Whole-cell patch-clamp pipettes and bath solutions were as described previously.20 Current density was calculated by dividing whole-cell current amplitude by cell capacitance (Cm). Cm was estimated by dividing the decay time constant of the capacitive transient in response to 5-mV hyperpolarizing voltage-clamp steps from ⫺40 mV by the series resistance (Rs). Adequate voltage control Cryosections (10-m thickness) perpendicular or parallel to the epicardial surface were made from the same area as the CV measurements. Perpendicular sections incubated with rabbit polyclonal anti-Cx43 antibody (Zymed) and mouse monoclonal anti-Ncadherin antibodies (Sigma) were obtained by the method described previously.21 Immunolabeled sections were mounted in Vectashield mounting medium (Vector Laboratories) and examined with a light microscope with epifluorescence equipment (Nikon Optiphot-2). Presence of fibrosis was analyzed from parallel sections fixed with 4% paraformaldehyde and stained with picrosirius red. Images from 6 randomly selected fields per section viewed with light microscopy were digitized, and the amount of fibrosis was analyzed with Image Pro Plus (version 5.02, Media Cybernetics). Computer Model We assessed L and T in linear strands of 100 longitudinally or transversally coupled subepicardial myocytes. Individual cells of the strands were described with the Luo-Rudy dynamic model of mammalian subepicardial ventricular myocytes (LRd model),22 including the transient outward current Ito1.23 As in other studies,24,25 entire cell length (for longitudinal conduction) or width (for transversal conduction) was used as the spatial discretization element in our computations, with elements connected by the lumped myoplasmic resistance (calculated from the myocyte dimensions and the myoplasmic resistivity of 150 ⍀/cm) and gap junctional resistance (calculated from the myocyte dimensions and the gap junctional resistivity). Action potential propagation was elicited by injection of an ⬇20% suprathreshold current pulse (duration 2 ms) into the first cell of the strand, at a frequency of 4 Hz. The activation wave of the sixth action potential was analyzed. We assumed that membrane ionic channel density per unit of membrane surface area did not change with increased cell size. We ran simulations at normal and reduced intercellular coupling with control and HF cell dimensions, assuming that cell shape was that of a rectangular box. CV and total AT per myocyte were calculated from the difference in AT of cell 20 and cell 80. Statistical Analysis Data are presented as mean⫾SEM. A 1-way ANOVA with the Student-Newman-Keuls test for post hoc analysis was used to test for a significant differences of L, T, TM, stimulation threshold, QRS duration, myocyte length and width, resting membrane potential, and sodium current density (either whole-cell or cell-attached patch clamp) in HF compared with control hearts. When data were not normally distributed or variance was not equal, a Kruskal-Wallis 1-way ANOVA on ranks with Dunn’s test for post hoc analysis was 808 Circulation TABLE 1. February 14, 2006 Functional Characteristics of Control and HF Rabbits Control (n⫽22) HF (n⫽44) BW, kg 4.3⫾0.1 4.2⫾0.1 HW, g 10.2⫾0.4 21.1⫾0.5* HW/BW, g/kg 2.4⫾0.1 5.0⫾0.1* LW/BW, g/kg 3.1⫾0.1 4.6⫾0.2* LVEDP, mm Hg 3.4⫾0.5 20.5⫾1.5* Ascites 0/22 21/44* QRS duration, ms 50⫾1 58⫾1* BW indicates body weight; HW, heart weight; HW/BW, relative heart weight; LW/BW, relative lung weight; and LVEDP, left ventricular end-diastolic pressure. Data are mean⫾SEM. *P⬍0.01 for HF vs control. BW comparisons are by ANOVA; HW, KruskalWallis; HW/BW, Kruskal-Wallis; LW/BW, Kruskal-Wallis; LVEDP, Kruskal-Wallis; ascites, 2; and QRS, ANOVA. Downloaded from http://circ.ahajournals.org/ by guest on June 18, 2017 used. This approach was used to test for significant differences of heart weight, lung weight, heart weight/body weight ratio, lung weight/body weight ratio, left ventricular end-diastolic pressure, and amount of fibrosis in HF compared with control rabbits. The presence of ascites in HF compared with controls was tested with the 2 test for statistical differences. P⬍0.05 indicates statistical significance. Results HF Parameters Table 1 shows characteristics of HF. There were 3 experimental groups: (1) measurement of CVs (control n⫽8; HF n⫽15), (2) myocyte dimensions (control n⫽6; HF n⫽7), and (3) patch-clamp experiments (control n⫽8; HF n⫽22). In Table 1, data from control (n⫽22) or HF (n⫽44) animals from each experimental group were lumped together because there were no significant differences between the parameters in the 3 subgroups. HF is present as evident from the presence of ascites and the increase in (relative) heart weight, relative lung weight, and left ventricular end-diastolic pressure compared with control. TABLE 2. CV in Control and HF Rabbits Control HF L, cm/s 67⫾4 (n⫽8) 79 ⫾2* (n⫽15) T, cm/s 37⫾2 (n⫽8) 43 ⫾2* (n⫽15) TM, cm/s 24⫾1 (n⫽8) 25 ⫾2 (n⫽10) Data are in centimeters per second. Data are mean⫾SEM. *P⬍0.01 for HF vs control. L: longitudinal CV (ANOVA); T: transversal CV (ANOVA); TM: transmural CV (ANOVA). control. This is also evident from the 15-ms isochrone (bold line), which encloses a larger area in the activation map of the HF than the control rabbit. The stimulation threshold was not different between HF (165⫾33 A) and control 164⫾25 A (ANOVA, P⫽NS). Average L and T (Table 2) were higher in HF than in control animals. TM was not different between control and HF animals (Table 2). Sodium Current Figure 2 shows representative whole-cell patch-clamp (Figure 2A) and cell-attached macropatch (Figure 2B) INa recordings from control and HF cells. Current density and current-voltage relationship (Figure 2B) were not different in HF compared with control in either condition (Figures 2C and 2D). Cx43 and Fibrosis Figure 3A shows normal Cx43 expression in all 3 layers of a control heart, which is accompanied by an almost identical pattern of N-cadherin expression (mechanical junction). Figure 3B shows a typical pattern of Cx43 in a heart from a rabbit with HF. Expression in the subendocardium (6/6 hearts) and the subepicardium (5/6 hearts) is similar to control. Cx43 (but also N-cadherin) in the midmyocardium is reduced (6/6 hearts). In 1 heart, epicardial Cx43 was reduced, although not to the level depicted for the midmyocardium. The total amount of fibrosis was not different between control (21⫾1%, n⫽4) and HF (20⫾2%, n⫽10; Kruskal-Wallis, P⫽NS). Myocyte Dimensions QRS duration (Table 1) was 58⫾1 ms (mean⫾SEM) in HF. This was longer than in the same animals before the first operation (49⫾1 ms) and in control animals (age 8 months; 50⫾1 ms; ANOVA, P⬍0.01). Myocyte length and width were measured in 6 control and 7 HF rabbits (50 myocytes/heart). Figure 4 shows that myocyte length (191.7⫾3.7 versus 144.9⫾2.4 m, respectively) and width (32.7⫾1.1 versus 25.2⫾0.5 m, respectively) were both increased by ⬇30% in HF compared with control (ANOVA, P⬍0.05). Conduction Velocity Simulation Studies Figures 1A and 1B shows representative subepicardial activation maps. Both L and T were higher in HF than in We performed computer simulations (Figure 5) to test whether the experimentally observed increase in myocyte QRS Duration Figure 1. Activation maps from a control (A) and HF (B) rabbit heart. Dots indicate individual electrodes on the multielectrode, which was placed on the left ventricular free wall. Lines indicate isochrones. Numbers indicate AT (in ms) relative to the time of stimulation. The increase in gray scale corresponds with the 5-ms spaced isochrones. In the white area, the AT ranges from 0 to 10 ms. Arrows indicate the direction and magnitude of L and T. Wiegerinck et al Conduction, QRS Width, and Heart Failure 809 Figure 2. Fast sodium current (INa) in left ventricular myocytes isolated from control and HF rabbits. A and B, Typical whole-cell (A) and cell-attached patchclamp (B) recordings of INa in response to a voltage clamp step from ⫺120 to ⫺30 mV. C and D, Average INa amplitude measured with (C) whole cell and (D) cell-attached patch clamp. Voltageclamp protocol shown as inset. See Methods for further details. C indicates control. Downloaded from http://circ.ahajournals.org/ by guest on June 18, 2017 dimensions (see Figure 4) can explain the HF-induced increase in L and T, as well as the increase in total AT as indicated by the increase in QRS duration. First, we ran simulations with cell length and width set to 144.9 and 25.2 m, respectively, ie, the mean values observed for control myocytes. We found that total gap junctional conductance values of 4 and 15 S were required to obtain the experimentally observed L and T, respectively. These values of 4 and 15 S correspond with a gap junctional resistivity (Rj) of 1.59 and 2.43 ⍀/cm2, respectively, and were regarded as 100% of intercellular coupling. Next, we ran simulations with cell length and width set to 191.7 and 32.7 m, respectively, ie, the mean values observed for failing myocytes. We assumed that the number of gap junction and membrane ionic channels per unit membrane surface did not change. Figure 6 shows simulated CV (Figures 6A and 6B) and total AT per myocyte (Figures 6C and 6D). Longitudinal CV (Figure 6A) increased from 68 cm/s (control) to 81 cm/s (HF) at 100% intercellular coupling; however, the 19% increase in L was not enough to fully compensate for the 32% increase in myocyte length, and therefore, total AT/myocyte (Figure 6C) was larger in HF than control (237 versus 213 s, 11% increase). Transversal CV (Figure 6B) was 37 cm/s in control and 47 cm/s in HF animals at 100% intercellular coupling. The 27% increase in T was large enough to almost completely compensate for the 30% increase in cell width. Total AT per myocyte in the strand of transversally coupled cells (Figure 6D) changed from 68 s in control to 70 s in HF animals (2.5% increase) at 100% intercellular coupling. To test the dependence of our simulation results on the degree of intercellular coupling, we repeated our simulations at 20%, 40%, 60%, and 80% intercellular coupling, respectively. Reduced coupling resulted in reduced CV, but at every identical simulated value for intercellular coupling, CV was larger in HF cells than in control cells. The dashed horizontal lines in Figures 6A and 6B show that the CV in HF cells with a coupling of 50% to 60% is similar to that of control cells with 100% intercellular coupling. Discussion In a nonischemic rabbit model of moderate HF compared with control, (1) QRS duration, L, T, and myocyte length and width are increased; (2) TM, sodium current density, fibrosis, and subepicardial Cx43 expression are unchanged; and (3) midmyocardial Cx43 expression is reduced. Computer simulations demonstrate that the increased cell size results in increased CV at any level of intercellular coupling; however, L and TM are not large enough to compensate for the increase in ventricular dimensions in HF and thus result in an increased QRS width. Figure 3. Representative immunohistochemical labeling of Cx43 and N-cadherin in left ventricle in control (A, n⫽6) and HF (B, n⫽6). Left, Cx43 expression; Middle, N-cadherin (N-cad) expression; Right, ensemble picture. Subendocardial (Endo), midmyocardial (Mid), and subepicardial (Epi) expression patterns are shown in the upper, middle and lower rows, respectively. Scale bar: 50 m. 810 Circulation February 14, 2006 Figure 4. Dimensions of isolated myocytes. A, Typical example of a myocyte from a control (left) and an HF (right) rabbit. B and C, Mean myocyte length (B) and width (C) in 6 control (white) and 7 HF (gray) rabbits (50 myocytes/rabbit). Downloaded from http://circ.ahajournals.org/ by guest on June 18, 2017 QRS Duration QRS prolongation occurs in patients with HF26 and is associated with increased cardiac mortality.4,5 In patients with dilated cardiomyopathy, QRS duration increases progressively.3,27 This indicates that even in the presence of complete bundle-branch block, increased left ventricular mass with increased CV may cause an increase in QRS duration, although it cannot be excluded that conduction is slowed in more severe HF. Indeed, in a study of individuals free of HF and myocardial infarction, QRS duration was positively correlated with left ventricular mass and dimensions.28 Sodium Current In cell-attached patches, INa was unchanged in HF compared with control animals (Figure 2B). This indicates that the brain-type sodium channels, located in T-tubules,29 do not Figure 5. Geometry of simulated strands of (A) longitudinally or (B) transversally coupled ventricular cells. Individual cells of the strand were described with the LRd model.22 Cell length (L) and width (W) were based on experimental data from left ventricular myocytes isolated from control or failing rabbit hearts (Figure 4). Rj denotes gap junctional resistivity. See Methods and text for further details. change in HF (Figure 2B). Whole-cell INa, carried by both myocardial-type (located in the intercalated disk30) and braintype sodium channels, is also not changed in HF (Figure 2A). Any change in CV is therefore attributable to changes in other parameters. In contrast, INa peak current density was decreased31 in other models. This may imply that patients with severe HF have a reduced CV that contributes to QRS prolongation. Intercellular Coupling and Fibrosis Reduction of Cx43 may contribute to slowing of conduction. Cx43 expression is reduced in the left ventricle of patients with hypertrophic, dilated, or ischemic cardiomyopathies.32–34 These studies did not specifically report on subepicardial Cx43 expression. In the present study, we observed reduced Cx43 expression in left ventricular midmyocardium but not in subepicardium. In contrast, in a canine model of rapid pacing–induced HF, Cx43 expression was reduced, especially in the subepicardial layer.35 However, in dogs with left bundle-branch block–related left ventricular dyssynchrony, the lateral wall showed no change in subepicardial Cx43 expression.36 Lower Cx43 expression levels in subendocardium compared with subepicardium were further exaggerated in dogs with left bundle-branch block associated with HF compared with controls.36,37 A transmural gradient of Cx43 expression was present in mice but not in rats.38 Apparently, the transmural gradient of Cx43 expression differs and changes within the heart, between species, and with the underlying disease. Tissue architecture and amount of fibrosis can affect activation propagation in the setting of HF39; however, fibrosis was not increased in the present model of HF. Structural remodeling (except for hypertrophy) was also absent because no infarction was made. Conduction Velocity In guinea pig after 150 days of aortic constriction, heart size, QRS duration,10 and intracellular resistivity40 were increased and CV11 was decreased. In a milder stage of HF (50 days of Wiegerinck et al Conduction, QRS Width, and Heart Failure 811 Figure 6. Simulation results obtained with the strands as described in Figure 5. A and B, Effect of myocyte dimensions on CV in strands of longitudinally (A) and transversally (B) coupled cells. The dashed line illustrates the conditions in which there is a balance between the effects of intercellular coupling and hypertrophy. C and D, Effect of myocyte dimensions on total AT per myocyte (TAT/myocyte) in strands of longitudinally (C) and transversally (D) coupled cells. Downloaded from http://circ.ahajournals.org/ by guest on June 18, 2017 aortic constriction), heart size and CV were increased,11 whereas intracellular resistivity40 and QRS width10 were unchanged. Other studies reported a decreased subepicardial CV in the setting of HF9 or no change.7,8 In the present animal model, we were able to directly relate CV to the increased cellular dimensions in subepicardium, where Cx43 was unchanged; however, this increased subepicardial CV (combined with unaltered TM) was not enough to compensate for the increase in myocyte dimensions and hence resulted in an increased QRS duration. The data obtained in guinea pig illustrate the dynamic nature of HF. In the present model, HF is moderate and electrical remodeling is absent, except for transient outward K⫹ current density (data not shown). This makes it an ideal model to address the effect of hypertrophy. In the present study, TM was not changed. In contrast, Toyoshima et al12 showed that TM, calculated by dividing the left ventricular wall thickness by the interval between onset of the QRS complex and the moment of epicardial breakthrough, is increased in hypertrophy, whereas in a canine model of rapid pacing–induced HF, TM was decreased.35 The discrepancy between these results might be due to differences in the amount of Cx43 expression or electrical remodeling. cell size is at least as important as Cx43 distribution in transversal propagation.41 The simulations in the present study show that the increase in T may compensate for the increased dimensions of the preparation, whereas the increase in L does not compensate for the increased length of the preparation and results in a longer total AT. Our simulations with subepicardial cells indicate that specific combinations of reduced intercellular coupling and increased myocyte size may yield an unchanged CV in HF, despite changes in both underlying parameters (cf the dashed lines in Figures 6A and 6B). This may also explain the unchanged TM despite reduced midmyocardial Cx43 levels in our rabbit model. Clinical Implications QRS prolongation may be associated with increased or normal CV in patients with HF/hypertrophy; however, the potential antiarrhythmic effect of increased CV in patients with HF/hypertrophy may be offset by a disproportional increase in myocardial mass, which favors the onset of reentrant arrhythmias. Study Limitations Myocyte Dimensions and Simulations In our computer model, axial resistivity is decreased owing to pathophysiological growth and results in increased CV. Increase in cell length in a similar model but one that compared adult with neonatal myocytes (wild-type and Cx43⫹/⫺ mice) also resulted in decreased axial resistivity and increased CV.25 In canine ventricle, normal physiological growth increased myocyte length and width and L and T in adult dogs compared with neonatal animals.41 The gap junction distribution also changed with aging. Computer simulations demonstrated that increase in myocyte size and Cx43 distribution are sufficient to account for the experimental results and that We directly compared CV of 2D measurements with 1D simulations. In the measurements, we used point stimulation, whereas our 1D simulations correspond with plane wave stimulation in 2D. Therefore, our measurements are subject to underestimation of planar CV due to wave-front curvature.42 Nevertheless, it is clear that the increase in cell size and CV are directly related. We used 1D simulations with rectangular boxlike cells, which does not reflect the mixed contribution of longitudinal and transverse gap junctions in interdigitating cell bundles. Length and width but not thickness of the myocyte could be measured with light microscopy. We used the width of the myocyte as an estimate for the thickness, but we cannot exclude an overestimation. 812 Circulation February 14, 2006 Our model of HF is relatively mild and focused on hypertrophy, although ascites is a predominant feature. The model permits study of increases in cell size and decreases in intercellular coupling. In more severe stages of HF, additional factors such as fibrosis or electrical remodeling also contribute to pathophysiological changes. A major cause of HF is ischemia related. We chose a model of combined volume/pressure overload to exclude infarcted tissue as a confounding factor. Studies in explanted hearts involve patients who were in the end stage of HF, whereas our model reflects a moderate stage of HF. Thus, conclusions derived from this particular model do not necessarily apply to other HF models. Conclusions Downloaded from http://circ.ahajournals.org/ by guest on June 18, 2017 QRS prolongation was associated with increased heart size and increased dimensions of left ventricular myocytes in our nonischemic rabbit model of moderate HF. Also, the animals with HF had higher left ventricular subepicardial CVs than control animals, without any change in sodium current density. Our computer simulations demonstrated that the increase in myocyte dimensions per se may explain the increase in subepicardial CV and the unchanged TM despite the reduction in midmyocardial Cx43 expression. Although subepicardial CV was increased and TM unchanged, this was not enough to compensate for increased heart size. This explains the increased QRS duration, as corroborated by our simulation study in which total AT was longer in longer strands with the same number of cells. Acknowledgments The authors thank Sara Tasseron, Shirley van Amersfoorth, and Diana Bakker for their analysis of fibrosis. The authors had full access to the data and take full responsibility for its integrity. All authors have read and agree to the manuscript as written. Disclosures None. References 1. Bigger JT Jr. Why patients with congestive heart failure die: arrhythmias and sudden cardiac death. Circulation. 1987;75:IV-28 –IV-35. 2. Poole-Wilson PA, Uretsky BF, Thygesen K, Cleland JG, Massie BM, Ryden L. Mode of death in heart failure: findings from the ATLAS trial. Heart. 2003;89:42– 48. 3. Xiao HB, Roy C, Fujimoto S, Gibson DG. Natural history of abnormal conduction and its relation to prognosis in patients with dilated cardiomyopathy. Int J Cardiol. 1996;53:163–170. 4. Bode-Schnurbus L, Bocker D, Block M, Gradaus R, Heinecke A, Breithardt G, Borggrefe M. QRS duration: a simple marker for predicting cardiac mortality in ICD patients with heart failure. Heart. 2003;89: 1157–1162. 5. Kearney MT, Zaman A, Eckberg DL, Lee AJ, Fox KA, Shah AM, Prescott RJ, Shell WE, Charuvastra E, Callahan TS, Brooksby WP, Wright DJ, Gall NP, Nolan J. Cardiac size, autonomic function, and 5-year follow-up of chronic heart failure patients with severe prolongation of ventricular activation. J Card Fail. 2003;9:93–99. 6. Mines GR. On circulating excitation in heart muscles and their possible relation to tachycardia and fibrillation. Trans R Soc Can. 1914;IV:43–52. 7. Pye MP, Cobbe SM. Arrhythmogenesis in experimental models of heart failure: the role of increased load. Cardiovasc Res. 1996;32:248 –257. 8. Zhu WX, Johnson SB, Brandt R, Burnett J, Packer DL. Impact of volume loading and load reduction on ventricular refractoriness and conduction properties in canine congestive heart failure. J Am Coll Cardiol. 1997; 30:825– 833. 9. Laurita KR, Chuck ET, Yang T, Dong WQ, Kuryshev YA, Brittenham GM, Rosenbaum DS, Brown AM. Optical mapping reveals conduction slowing and impulse block in iron-overload cardiomyopathy. J Lab Clin Med. 2003;142:83– 89. 10. Winterton SJ, Turner MA, O’Gorman DJ, Flores NA, Sheridan DJ. Hypertrophy causes delayed conduction in human and guinea pig myocardium: accentuation during ischaemic perfusion. Cardiovasc Res. 1994; 28:47–54. 11. Cooklin M, Wallis WR, Sheridan DJ, Fry CH. Conduction velocity and gap junction resistance in hypertrophied, hypoxic guinea-pig left ventricular myocardium. Exp Physiol. 1998;83:763–770. 12. Toyoshima H, Park YD, Ishikawa Y, Nagata S, Hirata Y, Sakakibara H, Shimomura K, Nakayama R. Effect of ventricular hypertrophy on conduction velocity of activation front in the ventricular myocardium. Am J Cardiol. 1982;49:1938 –1945. 13. Anderson KP, Walker R, Urie P, Ershler PR, Lux RL, Karwandee SV. Myocardial electrical propagation in patients with idiopathic dilated cardiomyopathy. J Clin Invest. 1993;92:122–140. 14. Vermeulen JT, McGuire MA, Opthof T, Coronel R, de Bakker JM, Klopping C, Janse MJ. Triggered activity and automaticity in ventricular trabeculae of failing human and rabbit hearts. Cardiovasc Res. 1994;28: 1547–1554. 15. Potse M, Linnenbank AC, Grimbergen CA. Software design for analysis of multichannel intracardial and body surface electrocardiograms. Comput Methods Programs Biomed. 2002;69:225–236. 16. Kléber AG, Janse MJ, Wilms-Schopmann FJ, Wilde AA, Coronel R. Changes in conduction velocity during acute ischemia in ventricular myocardium of the isolated porcine heart. Circulation. 1986;73:189 –198. 17. Kléber AG, Rudy Y. Basic mechanisms of cardiac impulse propagation and associated arrhythmias. Physiol Rev. 2004;84:431– 488. 18. Murray KT, Anno T, Bennett PB, Hondeghem LM. Voltage clamp of the cardiac sodium current at 37 degrees C in physiologic solutions. Biophys J. 1990;57:607– 613. 19. ter Welle HF, Baartscheer A, Fiolet JW, Schumacher CA. The cytoplasmic free energy of ATP hydrolysis in isolated rod-shaped rat ventricular myocytes. J Mol Cell Cardiol. 1988;20:435– 441. 20. de Groot JR, Veenstra T, Verkerk AO, Wilders R, Smits JP, WilmsSchopman FJ, Wiegerinck RF, Bourier J, Belterman CN, Coronel R, Verheijck EE. Conduction slowing by the gap junctional uncoupler carbenoxolone. Cardiovasc Res. 2003;60:288 –297. 21. van Veen TA, van Rijen HV, Wiegerinck RF, Opthof T, Colbert MC, Clement S, de Bakker JM, Jongsma HJ. Remodeling of gap junctions in mouse hearts hypertrophied by forced retinoic acid signaling. J Mol Cell Cardiol. 2002;34:1411–1423. 22. Faber GM, Rudy Y. Action potential and contractility changes in [Na⫹]i overloaded cardiac myocytes: a simulation study. Biophys J. 2000;78: 2392–2404. 23. Dumaine R, Towbin JA, Brugada P, Vatta M, Nesterenko DV, Nesterenko VV, Brugada J, Brugada R, Antzelevitch C. Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are temperature dependent. Circ Res. 1999;85:803– 809. 24. Shaw RM, Rudy Y. Ionic mechanisms of propagation in cardiac tissue: roles of the sodium and L-type calcium currents during reduced excitability and decreased gap junction coupling. Circ Res. 1997;81:727–741. 25. Thomas SP, Kucera JP, Bircher-Lehmann L, Rudy Y, Saffitz JE, Kleber AG. Impulse propagation in synthetic strands of neonatal cardiac myocytes with genetically reduced levels of connexin43. Circ Res. 2003; 92:1209 –1216. 26. Hombach V. Electrocardiogram of the failing heart. Card Electrophysiol Rev. 2002;6:209 –214. 27. Grigioni F, Carinci V, Boriani G, Bracchetti G, Potena L, Magnani G, Bacchi-Reggiani L, Magelli C, Branzi A. Accelerated QRS widening as an independent predictor of cardiac death or of the need for heart transplantation in patients with congestive heart failure. J Heart Lung Transplant. 2002;21:899 –902. 28. Dhingra R, Pencina MJ, Benjamin EJ, Levy D, Larson MG, Meigs JB, Rifai N, D’Agostino RB Sr, Vasan RS. Cross-sectional relations of urinary sodium excretion to cardiac structure and hypertrophy: the Framingham Heart Study. Am J Hypertens. 2004;17:891– 896. 29. Maier SK, Westenbroek RE, Schenkman KA, Feigl EO, Scheuer T, Catterall WA. An unexpected role for brain-type sodium channels in coupling of cell surface depolarization to contraction in the heart. Proc Natl Acad Sci U S A. 2002;99:4073– 4078. Wiegerinck et al 30. Cohen SA. Immunocytochemical localization of rH1 sodium channel in adult rat heart atria and ventricle: presence in terminal intercalated disks. Circulation. 1996;94:3083–3086. 31. Valdivia CR, Chu WW, Pu J, Foell JD, Haworth RA, Wolff MR, Kamp TJ, Makielski JC. Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J Mol Cell Cardiol. 2005;38:475– 483. 32. Dupont E, Matsushita T, Kaba RA, Vozzi C, Coppen SR, Khan N, Kaprielian R, Yacoub MH, Severs NJ. Altered connexin expression in human congestive heart failure. J Mol Cell Cardiol. 2001;33:359 –371. 33. Kostin S, Rieger M, Dammer S, Hein S, Richter M, Klovekörn WP, Bauer EP, Schaper J. Gap junction remodeling and altered connexin43 expression in the failing human heart. Mol Cell Biochem. 2003;242: 135–144. 34. Peters NS, Green CR, Poole-Wilson PA, Severs NJ. Reduced content of connexin43 gap junctions in ventricular myocardium from hypertrophied and ischemic human hearts. Circulation. 1993;88:864 – 875. 35. Poelzing S, Rosenbaum DS. Altered connexin43 expression produces arrhythmia substrate in heart failure. Am J Physiol Heart Circ Physiol. 2004;287:H1762–H1770. 36. Spragg DD, Akar FG, Helm RH, Tunin RS, Tomaselli GF, Kass DA. Abnormal conduction and repolarization in late-activated myocardium of dyssynchronously contracting hearts. Cardiovasc Res. 2005;67:77– 86. Conduction, QRS Width, and Heart Failure 813 37. Spragg DD, Leclercq C, Loghmani M, Faris OP, Tunin RS, Disilvestre D, McVeigh ER, Tomaselli GF, Kass DA. Regional alterations in protein expression in the dyssynchronous failing heart. Circulation. 2003;108: 929 –932. 38. Yamada KA, Kanter EM, Green KG, Saffitz JE. Transmural distribution of connexins in rodent hearts. J Cardiovasc Electrophysiol. 2004;15: 710 –715. 39. Kawara T, Derksen R, De Groot JR, Coronel R, Tasseron S, Linnenbank AC, Hauer RNW, Kirkels H, Janse MJ, De Bakker JMT. Activation delay after premature stimulation in chronically diseased human myocardium relates to the architecture of interstitial fibrosis. Circulation. 2001;104: 3069 –3075. 40. Cooklin M, Wallis WR, Sheridan DJ, Fry CH. Changes in cell-to-cell electrical coupling associated with left ventricular hypertrophy. Circ Res. 1997;80:765–771. 41. Spach MS, Heidlage JF, Dolber PC, Barr RC. Electrophysiological effects of remodeling cardiac gap junctions and cell size: experimental and model studies of normal cardiac growth. Circ Res. 2000;86:302–311. 42. Knisley SB, Hill BC. Effects of bipolar point and line stimulation in anisotropic rabbit epicardium: assessment of the critical radius of curvature for longitudinal block. IEEE Trans Biomed Eng. 1995;42: 957–966. Downloaded from http://circ.ahajournals.org/ by guest on June 18, 2017 CLINICAL PERSPECTIVE The width of the QRS complex is associated with the duration of complete activation of the ventricles. In hypertrophy and heart failure, QRS duration is increased. Generally, this increase is interpreted as a decrease in ventricular conduction velocity. We recorded QRS duration and measured conduction velocity (longitudinal, transversal, and transmural) in the left ventricle in an animal model of hypertrophy/heart failure. Additionally, we measured the size of left ventricular myocytes and studied the expression pattern of connexin43 in the left ventricle. QRS duration was increased in animals with heart failure compared with controls. Myocyte dimensions were increased. Connexin43 expression remained normal in subepicardium but was reduced in midmyocardium. Transmural conduction velocity was unaltered, and longitudinal and transversal subepicardial conduction velocities were increased. Computer simulations demonstrated that an increase in myocyte dimensions per se may explain the increase in subepicardial conduction velocity. Additionally, the increase in myocyte size in combination with the reduction in midmyocardial connexin43 expression may explain the unchanged transmural conduction velocity. Although subepicardial conduction velocity is increased and transmural conduction velocity unchanged, this is not enough to compensate for increased heart size, and hence QRS duration is increased. Therefore, a wide QRS complex, as observed, for example, in patients with heart failure, does not necessarily indicate slow ventricular conduction but can also be due to hypertrophy alone, which increases the total time required to activate the ventricles. Larger Cell Size in Rabbits With Heart Failure Increases Myocardial Conduction Velocity and QRS Duration Rob F. Wiegerinck, Arie O. Verkerk, Charly N. Belterman, Toon A.B. van Veen, Antonius Baartscheer, Tobias Opthof, Ronald Wilders, Jacques M.T. de Bakker and Ruben Coronel Downloaded from http://circ.ahajournals.org/ by guest on June 18, 2017 Circulation. 2006;113:806-813; originally published online February 6, 2006; doi: 10.1161/CIRCULATIONAHA.105.565804 Circulation is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231 Copyright © 2006 American Heart Association, Inc. All rights reserved. Print ISSN: 0009-7322. Online ISSN: 1524-4539 The online version of this article, along with updated information and services, is located on the World Wide Web at: http://circ.ahajournals.org/content/113/6/806 Permissions: Requests for permissions to reproduce figures, tables, or portions of articles originally published in Circulation can be obtained via RightsLink, a service of the Copyright Clearance Center, not the Editorial Office. Once the online version of the published article for which permission is being requested is located, click Request Permissions in the middle column of the Web page under Services. Further information about this process is available in the Permissions and Rights Question and Answer document. Reprints: Information about reprints can be found online at: http://www.lww.com/reprints Subscriptions: Information about subscribing to Circulation is online at: http://circ.ahajournals.org//subscriptions/