Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
J. CHEM. SOC. FARADAY TRANS., 1991, 87(4), 657-662
657
Magic-angle Spinning Nuclear Magnetic Resonance Studies of Water
Molecules adsorbed on Brginsted- and Lewis-acid Sites in Zeolites
and Amorphous Silica-Aluminas
Michael Hunger, Dieter Freude and Harry Pfeifer
Sektion Physik der Universitat Leipzig, Linnestrafle 5 , 0-7010 Leipzig, Germany
On weakly rehydrated dealuminated zeolites and amorphous silica-aluminas the presence of different types of
acidic centres can be ascertained by proton magic-angle spinning nuclear magnetic resonance ('H MAS NMR)
measurements. A 'H NMR line at ca. 6.5ppm is caused by water adsorption on Lewis-acid sites. The shift of the
'H NMR line of Brernsted-acid sites (bridging OH groups) to lower field for hydrated samples can be interpreted
quantitatively by a fast proton exchange between water molecules, bridging OH groups and hydroxonium ions.
Zeolites and amorphous silica-aluminas, which are denoted
in the following as aluminosilicates, may contain
Brernsted-acid sites, known to be bridging hydroxyl groups
(SiOHAl), and also Lewis-acid sites. If small amounts of
water are adsorbed, the water molecules can be involved in
the formation of hydroxonium ions. They can be dissociated
or strongly adsorbed by Lewis-acid sites.
Vega and Luz'-~ studied zeolites of type H-RHO by 'H
MAS NMR. They found after addition of small amounts of
water on activated samples a narrow peak at 5.9 ppm and a
broad signal at ca. 4 ppm, while at the highest loading the
spectrum consists of two narrow peaks at 4.6 and 9.1 ppm.
Only the signal at 4.6 ppm could be unambiguously identified
with water physically adsorbed in the zeolite cavities. Bronnimann et aL4 found three narrow lines in the 'H MAS NMR
spectra of weakly hydrated aluminosilicates and assigned the
signal at 7.0 ppm to hydrated Brernsted-acid sites. Hence a
bare Brernsted-acid site should have a much higher chemical
shift. In a previous paper on hydrated H-ZSM-5 zeolites
Scholle et aL5 assigned a broad line at 6 ppm to protons of
Bronsted-acid sites exchanging rapidly with protons of the
water molecules adsorbed on these sites. In our first attempt
to study the acidic properties of aluminosilicates by 'H MAS
NMR at a low field6 we assigned the line at 7 ppm to bridging hydroxyl groups. This interpretation was revised by the
statement7 that this line is caused by fast proton exchange
between residual ammonium ions and the bridging hydroxyl
groups. Through a direct observation of the bare bridging
OH groups by a measurement of evacuated samples at high
field and using CH, as an internal standard we could show
unambiguously that the chemical shift of these Brernsted-acid
sites in zeolites is between 3.8 and 5.2 ppm relative to
TMS.8-'o The exact value depends on the Si/Al ratio of the
zeolite framework and on the location of the hydroxyl group
within this framework. This result is in accordance with ref.
11 and 12. In a more recent paper13 we assigned a line at
6.5 ppm which appears in the spectra of hydrothermally pretreated and activated H-ZSM-5 zeolites after partial
rehydration to water molecules interacting with Lewis-acid
sites. Until now the 'H NMR signal of hydroxonium ions
could not be observed on hydrated aluminosilicates, although
Jentys et al.14 were able to assign two IR bands at 2457 and
2885 cm-' in the spectra of hydrated H-ZSM-5 to hydroxonium ions. Akitt" gives a value of 19.2 +_ 0.2 ppm for the
chemical shift of hydroxonium ions in various aqueous acids.
measured values of 10.7 and 11.4 ppm for
Ratcliffe et
hydroxonium protons in H,OClO, and on K + positions in
alunite, respectively. More recent results of ab initio calculations lie in the range 7.35-9.5 ppm for hydroxonium ions
without hydrogen bonding. 1 6 , ' In conclusion, for the 'free'
hydroxonium ion a chemical shift of 7-12 ppm is expected. A
partially hydrogen-bonded hydroxonium ion should appear
in the middle of the range 7.35-19.2 ppm, i.e. at ca. 13 ppm.
The affinity of a water molecule to combine with other
species can be estimated by a comparison of the values of ab
initio energies calculated by Sauer et al. :18*' H,0-A13 +,
791 kJ mol-'; H20-Mg2+, 335 kJ mol-'; H,O-Na',
117
kJ mol-'; H,O-SiOHAl, 58.4 kJ mol-'; H,O-H,O,
20.1
kJ mol-'; H,O-SiOH, 16.4 kJ mol-'. Therefore, the hydration of aluminosilicates should start with the adsorption of
water molecules on Lewis-acid sites (extraframework aluminium species and cations) followed by an adsorption on
bridging O H groups with the formation of hydroxonium
ions. Apparently, water molecules adsorbed on Lewis-acid
sites and hydroxonium ions exchanging their protons rapidly
with water may therefore contribute to a signal at 4-7 ppm in
the 'H MAS NMR spectra. It is the aim of the present paper
to demonstrate that Brransted- and Lewis-acid sites of weakly
hydrated aluminosilicates give rise to a signal in this range
and to show how they can be distinguished. The following
samples were under study : aluminosilicate, a hydrothermally
dealuminated H-Y zeolite denoted as De-Y and a shallowbed activated H-Y zeolite. In addition, two-pulse spin-echo
experiments combined with magic-angle spinning of the
samples were performed to study the proton exchange among
the various water species during the time interval between the
two pulses.
Experimental
Sample Preparation
The preparation of the aluminosilicate (15 wt.% Al,O,) was
described in ref. 6. After ion exchange by a 1 mol dm-3
aqueous solution of ammonium sulphate, the concentration
of sodium was <0.02 wt.%. The ammonium form of the Y
zeolite was prepared by cation exchange of 88 & 3% in an
aqueous solution of ammonium nitrate starting from an
Na-Y zeolite (Chemie AG Bitterfeld-Wolfen) with an Si/Al
ratio of 2.6. The hydrothermal treatment was carried out in a
tube of 5 mm i.d. with 8 mm maximum bed depth containing
ca. 2 g of the H-Y zeolite. At first the temperature was
increased up to 810 K at a rate of 10 K min-' in a water-free
nitrogen stream flowing at 1 dm3 min-'. The sample was
then steamed at this final temperature for 20 h under a
vapour pressure of 4 kPa, adjusted by the temperature of the
water bath through which the nitrogen was flowing.
For the 'H MAS NMR measurements all samples were
subjected to a shallow-bed activation procedure (670 SB). At
J. CHEM. SOC. FARADAY TRANS., 1991, VOL. 87
658
first the temperature was increased under vacuum at a rate of
10 K h- '. The samples were then kept at the final activation
temperature of 670 K at a pressure
Pa for 24 h and
finally cooled to room temperature and sealed. Before being
sealed, the samples were loaded at room temperature under
vacuum with measured amounts of doubly distilled water.
For the 27Al NMR measurements quartz tubes of 8 mm i.d.
were used instread of glass.
All 27Al MAS NMR measurements for the determination
of the framework Si/Al ratio were carried out on fully
rehydrated samples.
NMR Measurements
Measurements were made on a Bruker MSL 300 spectrometer. The home-made MAS equipment for the rotation of the
fused glass ampoules was carefully cleaned to avoid spurious
proton signals. A Larmor frequency of 300 MHz, a pulse
length of 4 ps for the 4 2 pulse, a recycle delay of 10 s and a
rotational frequency of 2500 f 5 Hz were used for the 'H
MAS NMR and spin-echo experiments. The 27Al NMR
pulse length was 1 p, which corresponds to a 4 1 2 pulse for
non-selective excitation.
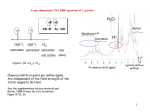
water an additional signal at 6.5 f 0.2 ppm appears [Fig. l(b)
and (c)], which can be assigned to water adsorbed on Lewisacid sites. The resonance position (6,) of this line does not
depend on the amount of adsorbed water up to 1 x 10''
molecules g- (cL). Although for the fully hydrated sample
(ca. 20 x 10'' H 2 0 g-') the 'H MAS NMR spectrum is
dominated by a broad signal at 4.8 f 0.4 ppm due to physically adsorbed water, the signals at 1.8 and 6.5 ppm are still
present as shoulders [Fig. l(d)]. The 27Al MAS NMR spectrum of the fully hydrated aluminosilicate in Fig. l(e) shows
only one line at 60 ppm due to tetrahedrally coordinated aluminium atoms.
Fig. 2 shows 'H MAS NMR spectra of the hydrothermally
dealuminated H-Y zeolite (De-Y) with a framework Si/Al
ratio of 5.0 f 0.5. The spectrum of the dehydrated sample
[Fig. 2(a)] consists of two signals, a peak at 1.8 f 0.2 ppm
due to 17 f 3 silanol groups per unit cell, on the external
surface of the crystallites and at crystal defects, and a shoulder at 4.0 f 0.2 ppm due to 5.6 f 1.5 bridging OH groups
per unit cell. As shown in Fig. 2(b), after the adsorption of a
few water molecules per unit cell, a line at 6.5 f 0.2 ppm (6,)
appears, which has weak MAS sidebands (<20% of the
whole intensity of the signal at 6.5 ppm) and does not shift up
to a loading of 10 H 2 0 per unit cell [Fig. 2(c)]. The
'
Results
Fig. l(a) shows the 'H MAS NMR spectrum of the dehydrated aluminosilicate, which consists, in agreement with a
previous study,20 of two signals due to silanol groups and
bridging OH groups, giving rise to a peak at 1.8 & 0.2 ppm
with a shoulder at 4.0 f 0.3 ppm. Spectral deconvolution of
signals including sidebands and comparison with a standard
yields (5.5 f 1.0) x lo2' silanol groups and (2.6 f 1.0) x lo2'
bridging OH groups g-', respectively. After loading with
h
-
I
I
60
0
47A,
60
O
4 7 * , (PPm)
Fig. 1 'H MAS NMR spectra of aluminosilicate, activated under
670SB conditions: unloaded (a) and after weak (b), (c) and full (d)
rehydration ( G l x 10'' and ca. 20 x 10'' H 2 0 g-', respectively)
and the 27Al MAS NMR spectrum of sample (6) (e) (*, MAS
sidebands)
(PPm)
Fig. 2 'H MAS NMR spectra of the hydrothermally dealuminated
H-Y zeolite (De-Y), activated under 670SB conditions : unloaded (a),
rehydrated with up to 10 H,O per unit cell (b) and (c), ca. 40 H 2 0
per unit cell (d) and after full (ca. 240 H,O per unit cell) rehydration
(e) and 27Al NMR and 27Al MAS NMR spectra of samples (d)and
(e) [(f)and (g), respectively] (*, MAS sidebands)
J. CHEM. SOC. FARADAY TRANS., 1991, VOL. 87
659
maximum concentration of water molecules giving rise to this
line is 2 H 2 0 per unit cell (cL).The shoulder due to bridging
OH groups is not aEected by a small loading [Fig. 2(b)].
With a higher loading a broad line at ca. 4 ppm appears
which is caused by physically adsorbed water molecules [Fig.
2(d)]. After full rehydration (240 H 2 0 per unit cell) the last
signal becomes narrower due to enhanced mobility of the
adsorbed water molecules and Its position is shifted to ca. 5
ppm, which agrees approximately with the value for bulk
water (4.8 k 0.2 ppm). In addition, a weak signal at 9.2 0.2
ppm with an intensity of 13.8 & 1 H 2 0 per unit cell due to
2.3 k 0.2 aluminium hexaaquo complexes2' per unit cell can
be observed [cf: Fig. 2(e) and Table 11. No 27Al NMR signal
appears for the dehydrated De-Y zeolite, in agreement with
previous studies.22 The 27Al NMR spectrum of the weakly
rehydrated sample (40 H 2 0 per unit cell) shows a broad line
with the maximum at ca. 60 ppm and a small shoulder at ca.
0 ppm, which can be assigned to tetrahedrally coordinated
framework aluminium atoms and aluminium hexaaquo complexes [Al(H,O);+], respectively [Fig. 2(f')]. The 27Al MAS
NMR spectrum of the fully rehydrated De-Y zeolite [Fig.
2(g)] consists of a line at 60 ppm, yielding a concentration of
32 k 2 framework aluminium atoms per unit cell and a signal
at 0 ppn, which corresponds to 6.4 k 0.6 AI(H20)2+ per unit
cell.
two-pulse spin-echo
According to Olejniczak et
experiments in rotating solids require pulse delays, z, which
are multiples of the MAS period to refocus the Hahn echo at
times of rotational echoes. Fig. 3 shows 'H MAS NMR
spectra obtained by a Fourier transform of the decaying part
of the spin echoes measured for the De-Y zeolite rehydrated
with 40 H 2 0 per unit cell and values for the pulse delay t of
1 ps [Fig. 3(a)], 400 ps [Fig. 3(&)], 1.6 ms [Fig. 3(c)] and 3.2
ms [Fig. 3(d)]. The spectrum recorded with z = 1 ps is identical to the spectrum shown in Fig. 2 ( 4 For the pulse delay
z = 400 ps the intensity of the broad signal due to physically
adsorbed water molecules which appears at ca. 4-5 ppm is
reduced drastically and can be ascertained only as a small
shoulder. The line at 6.5 0.2 ppm disappears only at much
greater values of the pulse delay [z = 3.2 ms, Fig. 3(d)]. For a
smaller loading of this zeolite (2 H 2 0 per unit cell) the signal
at 6.5 0.2 ppm can be observed even for z = 3.2 ms [Fig.
3(e)]. Analogously recorded spectra without application of
MAS show no thermally narrowed signal at 6.5 ppm.
Table 1 Values for the concentration and for the chemical shift of
hydroxyl groups and aluminium species in H-Y and De-Y zeolites,
determined by 'H MAS NMR and 27AIMAS NMR spectroscopy
H-Y
(PP~)
cSiOH
(per unit cell)
bSiOH
6SiOHAI
brn)
cSiOHAl
(per unit cell)
6' (PPm)
cL(per unit cell)
'AI(H20)63+ (ppm)
C A I ( H z 0 ) 6 3 + (per unit cell)
'H MAS NMR
1.8 f 0.2
2.4 & 0.4
4.0 f 0.2; 5.0 & 0.2
46 & 4
*
*
9.2 f 0.2
1.4 & 0.2
MAS NMR
60&2
52 f 2
2.7 & 0.1
De-Y
1.8 & 0.2
17k3
4.0 f 0.2
5.6 f 1.5
6.5 +_ 0.2
2.0 & 0.5
9.2 & 0.2
2.3 f 0.2
2 7 ~ 1
(ppm)
cAIF(per unit cell)
Si/AIF
cA,EF(per unit cell)
CAI(HzO)63 (per unit cell)
sAIF
*
3.4 & 0.3
60+2
32 f 2
5 & 0.5
20 k 4
6.4 f 0.6
*, Not observed. The concentration of residual sodium cations is
6.2 f 0.2 N a + per unit cell. F denotes framework and EF extraframework.
*
/
I
9.26.5I 1.8
4.8
6IH(PPm)
Fig. 3 ' H MAS NMR spectra obtained by a Fourier transformation of the spin echo measured for De-Y zeolite which was activated
under 670SB conditions and rehydrated with ca. 40 H,O per unit
cell. The MAS frequency was 2500 5 Hz and the values for the
pulse delay z were chosen as 1 ps (a), 400 ps (b), 1.6 ms ( c ) and 3.2 ms
(d). (e) was measured for a rehydration of 2 H,O per unit cell and a
pulse delay of z = 3.2 ms (*, MAS sidebands)
The 'H MAS NMR spectrum of the dehydrated H-Y
zeolite [Fig. qa)] consists of four overlapping signals, which
are broadened by proton-proton dipolar interaction (cf: ref.
10): a signal at 1.8 k 0.2 ppm corresponding to 2.4 k 0.4
SiOH groups per unit cell, signals at 4.0 k 0.2 and 5.0 +_ 0.2
ppm caused by bridging O H groups (SiOHAl) pointing
towards the zeolite supercages ( 0 1 atoms) and into the sodalite cages ( 0 3 atoms), respectively, and a signal at ca. 7 pprn
due to 0.3 & 0.1 residual NH: cations per unit cell.
The intensity of the bridging OH groups was determined
by deconvolution of the spectrum into Gaussian components,
including the spinning sidebands. The total concentration of
bridging OH groups is 46 & 4 per unit cell, in good agreement with a value determined in a previous study.' In Fig.
4(b)-(e) 'H MAS NMR spectra are shown for the H-Y zeolite
with an increasing loading with water. The maximum value
[Fig. 4(e)] corresponds to 50 water molecules per unit cell, i.e.
ca. one water molecule per bridging O H group. A fully
hydrated H-Y zeolite [Fig. qf)]contains ca. 240 water molecules per unit cell. Subtracting the weak signal of silanol
groups, the chemical shift 6, of the centre of gravity of the 'H
MAS NMR signal depends on the water content as follows
[Fig. 4(aHe)]: 6, = 4.3 ppm for 0 H 2 0 per unit cell, 6, = 4.6
ppm for 20 H,O per unit cell, 6, = 5.2 ppm for 30 H 2 0 per
unit cell, 6, = 5.8 ppm for 43 H,O per unit cell and 6, = 6.2
ppm for 50 H 2 0 per unit cell. The 'H MAS NMR spectrum
of the fully rehydrated H-Y zeolite [Fig. 4(f)] shows, similarly to Fig. 2(e), as well as the signal of physically adsorbed
water molecules at ca. 5.0 ppm a line of aluminium hexaaquo
complexes at 9.2 & 0.2 ppm. The intensity of this signal corresponds to 1.4 & 0.2 Al(H2O):+ complexes per unit cell. In
contrast to the 27AlNMR spectrum of the weakly rehydrated
H-Y zeolite (50 H 2 0 per unit cell), which shows only one
broad line at ca. 60 ppm [Fig. 4(g)], the 27A1 MAS NMR
J. CHEM. SOC. FARADAY TRANS., 1991, VOL. 87
660
water molecules adsorbed on Bronsted-acid sites (SiOHA1)
and physically adsorbed water molecules (H20-H,O).
The fact that for an equal amount of water molecules and
bridging hydroxyl groups only a single broad line appears
[cf Fig. qe)] must be explained by a fast proton exchange.
Previouslyz4 a value of 80 ps for the mean lifetime of protons
in bridging O H groups of partially hydrated H-Y zeolites at
300 K was determined. With increasing water loading the
intensity of the spinning sidebands decreased (cf. Fig. 4).
According to theory," this is caused by an enhancement of
the proton mobility with increasing water loading. In
shallow-bed activated H-Y zeolites the dependence of the 'H
NMR shift on water loading found in the present paper
agrees with the result of Mastikhin and Lapina,,' who
explained the resonance shift of 1 ppm, going from ca. 10 to
ca. 90 H 2 0 per unit cell, by a fast proton exchange between
bridging O H groups, water molecules and hydroxonium ions.
In principle one should also consider an exchange with water
molecules hydrogen bonded to bridging O H groups. These
species, however, could not be observed in the IR spectra of
rehydrated H-ZSM-5 zeolites.l 4 The proton magnetic resonance shift 6, can be described quantitatively by the equation:
A
Fig. 4 'H MAS NMR spectra of H-Y zeolite, activated under
670SB conditions: unloaded (a), rehydrated with up to 50 H,O per
unit cell or 1 H,O per bridging OH group [(bHe)]and fully (ca. 240
H,O per unit cell) rehydrated (f)and 27A1 NMR and 27Al MAS
NMR spectra of samples (e) and (f)[(g) and (h), respectively] (*,
MAS sidebands)
spectrum of the fully rehydrated H-Y sample [Fig. q h ) ] consists of a sharp signal at 60 ppm corresponding to a concentration of 52 & 2 framework aluminium atoms per unit cell
and a line at 0 ppm due to 3.4 f 0.3 A1(H20)2+ complexes
per unit cell. The absence of an 27Al NMI? signal of aluminium hexaaquo complexes in the spectrum of H-Y zeolite
loaded with 50 H,O per unit cell shows that the A13+
species, which cause these complexes in the fully rehydrated
sample, are formed only at higher degrees of rehydration by a
solution of extraframework aluminium species or by a
removal of framework aluminium atoms. In agreement with
this finding, in the dehydrated H-Y zeolite the sum of the
concentrations of bridging O H groups (46 f 4 SiOHAl per
unit cell) and residual sodium cations (6.2 & 0.2 Na+ per unit
cell) is equal to the concentration of framework aluminium
atoms (52 f 2 AIF per unit cell) within the limits of error (cf
Table 1).
Discussion
Taking into consideration the affinity of a water molecule
with different adsorption sites in the hydrogen form of
aluminosilicates, the following species must be discussed :
water molecules adsorbed on Lewis-acid sites (e.g. A],+),
where N i denotes the proton concentrations and di the
chemical shifts with i = 1 for bridging hydroxyl groups, i = 2
for physically adsorbed water molecules and i = 3 for hydroxonium ions. The following values have been used for the
chemical shifts (ppm): 6, = 4.3, 6, = 3.2-4.8 (3.2 for physisorbed water on silica gel2' and 4.8 for hydrogen-bonded
waterz6), and 6, = 13. From the value of the chemical shift
6, = 6.2 ppm measured for the sample loaded with 50 H,O
per unit cell and the values of 6,, 6, and 6, given above, a
concentration of 8.9-13.7 hydroxonium ions per unit cell
results. Hence, the probability of finding a water molecule in
the state of a hydroxonium ion in H-Y zeolite hydrated with
one water molecule per bridging O H group is ca. 0.2-0.3.
The nature of the extraframework aluminium species in the
hydrothermally treated and dehydrated H-Y zeolite (denoted
De-Y) is still unknown.27No information can be provided by
27Al MAS NMR, since the signals are strongly broadened by
quadrupole interaction and, therefore, all aluminium is NMR
invisible. By chemical methods oligorneric and polymeric
rather than monomeric extraframework aluminium species
have been found in hydrothermally dealuminated H-Y zeolites.28 Deeply dealuminated zeolites contain equal concentrations of framework aluminium atoms and bridging
hydroxyl groups (cf. ref. 8). Consequently, the amount of
charged extraframework aluminium species in strongly
hydrothermally dealuminated H-Y zeolites must be small.
The weak hydrothermal treatment of De-Y zeolite used in
the present study (4 kPa water vapour pressure) causes a
removal of 20 f 4 A1 per unit cell from framework positions.
The residual concentration of 32 f 2 A1 per unit cell on
framework positions is much higher than the concentratioo
of 6.2 f 0.2 Na+ cations and 5.6 & 1.5 bridging O H groups
per unit cell (see Table 1). Therefore, the De-Y zeolite must
contain (32 f 2) - (6.2 & 0.2) - (5.6 & 1.5) ;2: 20 f 4 positive
charges per unit cell on extraframework species.
determined
In a dealuminated H-Y zeolite Maher et
14.7 aluminium atoms per unit cell on cationic SI' positions.
In contrast to the value of 20 f 4 extraframework species per
unit cell (determined for the De-Y zeolite) which are supposed to be adsorption sites for water molecules, we observed
a maximum concentration of only two strongly adsorbed
water molecules per unit cell at an 'H WMR shift of 6.5 ppm
J. CHEM. SOC. FARADAY TRANS., 1991, VOL. 87
in the range of adsorption up to 40 H 2 0 per unit cell. In Fig.
2 ( 4 the broad line at a position corresponding to the 'H
NMR shift of bulk water is a hint that, apart from the abovementioned sites adsorbing strongly two water molecules per
unit cell, these ca. 20 charged extraframework species per unit
cell of De-Y zeolite do not act as additional adsorption sites
or cause a dissociation of water molecules.
Because of the large bonding energy of water molecules
adsorbed on A13+ cations (791 kJ mol-', see Introduction),
the appearance of the 'H MAS NMR signal at 6.5 ppm in
the spectra of aluminosilicates and dealuminated H-Y zeolite
(De-Y) has to be discussed in relation to the existence of
cationic extraframework aluminium species. At first it is
important to note that there is no evidence in the 'H MAS
NMR spectra of aluminosilicate and De-Y zeolite for the dissociation of water on cationic sites, since no increase of the
concentration of bridging hydroxyl groups could be observed
upon rehydration in contrast to the behaviour of the
alkaline-earth-metal cation-exchanged Y zeolite." In the
fully hydrated dealuminated H-Y zeolite aluminium hexaaquo complexes exist, and these give rise to a signal at ca. 9
ppm and at 0 ppm in the 'H MAS NMR and 27Al MAS
NMR spectra, respectively. The same can be found also for
the fully hydrated H-Y zeolite (Fig. 4). In contrast, the
aluminosilicate studied in the present paper shows neither in
the 'H MAS NMR nor in the 27AlMAS NMR spectra of the
fully rehydrated sample signals of aluminium hexaaquo complexes. With respect to the aluminium hexaaquo complexes
observed in the H-Y and De-Y zeolites it is of interest to note
that their concentration determined from the line at ca. 9
ppm in the 'H MAS NMR spectra is lower than the concentration determined from the line at 0 ppm in the 27Al MAS
NMR spectra. According to Table 1 the former value is
smaller by a factor of ca. 2-3. This result can be explained by
assuming that only for those aluminium hexaaquo complexes
which give rise to a separate line in the 'H MAS NMR
spectra is the proton exchange with the other water molecules sufficiently slow (exchange rate <ca. lo3 s-l). Presumably this may be the case for aluminium hexaaquo complexes
adsorbed in the small cages of the zeolite.
In order to throw some light upon the nature of the sites
which give rise to the signal at 6.5 ppm, two-pulse (Hahn's)
spin-cho experiments were performed. It is well known that
the spin echo appearing after two rf pulses (cf: ref. 31) is
reduced in amplitude if during the time interval 22 between
the first rf pulse and the echo, the local magnetic fields acting
upon the spins change. From Fig. 3(e) it can be seen that for
the De-Y zeolite rehydrated with 2 H 2 0 per unit cell the
water molecules which cause the signal at 6.5 ppm must stay
at the respective Lewis-acid sites for the order of 6 ms or
longer. The disappearance of the 'H NMR signal at 6.5 ppm
for the same zeolite and the same value of the pulse delay
after rehydration with 40 H 2 0 per unit cell [Fig. 3(d)] has to
be explained by a decrease of the residence time to values < 6
ms. On the other hand, the appearance of the line at 6.5 ppm
in the 'H MAS NMR spin-echo spectrum recorded with a
pulse delay of z = 1.6 ms [Fig. 3(c)] shows that the value of
the residence time must be of the order of 3 ms or larger,
yielding an estimate of 4 ms.
In the following we shall use this result to interpret quantitatively the sideband pattern of the 'H NMR signal at 6.5
ppm. Assuming that this signal is caused by water molecules
adsorbed on extraframework A13 cations with a mean
residence time z, between two statistical jumps of the water
molecule, the envelope of the free-induction decay can be
shown" to be given by the following:
+
@(tj= exp( - (w2/3)[2Z(R)
+ I(2R)l)
66 1
where
I(R) = tz,(l
+ Q2z,2)- ' + zC2(R22,2- 1x1 +
$222;)-
*
x [1 - exp( - t/z,)cos(Rt)]
+
- 2 0 ~ 3 1 n22:j-
exp( - t/z,)sin(Ot).
R denotes the spinning frequency of the sample and ( w ' ) the
second moment of the magnetic dipolar interaction. As a
lower limit for this quantity we use the formula for the
proton-aluminium magnetic dipolar interaction :
(a2)= 0.902 x 105/riA,
with ( w 2 ) in s P 2 and the proton-aluminium distance rHAl
in nm (cf: ref. 32). By an ab initio calculation Sauer" has
found a value of 0.176 nm for r A I O . [See (l)]
Using the value for the 0-H distance and the H-0-H
angle a in a water molecule of 0.0958 nm and 104.45", respect i ~ e l y rHAI
, ~ ~can be calculated to be 0.239 nm.
In Fig. 5 'H MAS NMR spectra computed by a Fourier
transformation of @(t) are shown for a spinning frequency R
of 2.5 kHz, a correlation time z, of 4 ms (as estimated above
for the signal at 6.5 ppm of weakly rehydrated De-Y zeolite)
and various values of the second moment of the H-A1 magnetic dipolar interaction corresponding to H- A1 distances
between 0.2 and 0.3 nm. In contrast to the experimentally
observed 'H NMR signal at 6.5 ppm [see Fig. 2(b)] the theoretical spectrum computed for the most probable distance
rHAl of 0.239 nm (which corresponds to rAIO= 0.176 nrnl8)
shows relative intensities of the MAS sidebands of > 50%.
Taking into consideration that the second moment may be
even larger owing to the intramolecular dipolar proton-proton interaction the discrepancy is even higher. This fact
and also the above-mentioned result that for the weakly
hydrated zeolites and aluminosilicate a signal at 6.5 ppm
appears but that after full hydration (for the zeolites only)
aluminium hexaaquo complexes can be observed, leads to the
conclusion that the signal at 6.5 ppm is not related to extraframework A13 species.
+
f
wo
wo
+2R + R
wo
w o u'o
- R -2R
Fig. 5 'H MAS NMR spectra computed for a spinning frequency
of 2.5 kHz, a correlation time z, of 4 ms as and various values of the
second moment of the Al-H magnetic dipolar interaction corresponding to H-A1 distances rHA, between 0.2 and 0.3 nm (rAl0
between 0.133 and 0.239 nm)
J. CHEM. SOC. FARADAY TRANS., 1991, VOL. 87
662
Through a quantitative analysis of 'H MAS NMR and
MAS N M R spectra22 obtained for H-Y zeolites activated at temperatures between 570 and 770 K under deepbed conditions it could be shown that the concentrations of
bridging OH groups and framework aluminium atoms
decrease simultaneously to the same extent. Therefore, weI9
suggest a model of a combined dehydroxylation of one bridging OH group and removal of one aluminium atom from the
framework. A two-step model for the dehydroxylation of
bridging OH groups has been discussed by Kuh134 [see (2)]
where in the first step threefold coordinated atoms of aluminium and of positively-charged silicon are formed.
5 K. F. M. G. J. Scholle, A. P. M. Kentgens, W. S. Veeman, P.
Frenken and G. P. M. van der Veiden, J Am. Chem. SOC., 1984,
88, 5.
6 M. Hunger, D. Freude, H. Pfeifer, H. Bremer, M. Jank and K-P.
Wendlandt, Chem. Phys. Lett., 1983, 100, 29.
7 H. Pfeifer, D. Freude and M. Hunger, Zeolites, 1985, 5, 274.
8 D. Freude, M. Hunger and H. Pfeifer, Z . Phys. Chem. N F , 1987,
152, 171.
9 M. Hunger. D. Freude, T. Friihlich, H. Pfeifer and W. Schwieger, Zeolites, 1987,7, 108.
10 H. Pfeifer, J. Chem. Soc., Faraday Trans. I, 1988,84,3777.
1 1 V. M. Mastikhin and K. I. Zamaraev, Z . Phys. Chem. N F , 1987,
152, 59.
12 G. Engelhardt, H-G. Jerschkewitz, U Lohse, P. Sarv, A.
Samoson and E. Lippmaa, Zeolites, 1987,7, 289.
H
H
AIO'
13 M. Hunger, D. Freude and H. Pfeifer, Caial. Today, 1988,3, 507.
I
I
14 A. Jentys, G. Warecka, M. Derewinski and J. A. Lercher, J.
\
o\ ?\
?\ ?\ 9-20 9Si Al Si?\Al +Si? 9Si?\-?\
Phys. Chem., 1989,93,4837.
Al Si
Si
Si Al Si Al Si
/ \ / \ / \ / \ / \
/ \ / \ / \
/ \
/ \ / \ / \ / \ / \
15 J. W. Akitt, J. Chem. Soc., Dalton Trans., 1973,49.
16 C. I. Ratcliffe, J. A. Ripmeester and J. S. Tse, Chem. Phys. Lett.,
1985,120,427.
17 J. Sauer, J. Mol. Catal., 1989, 54, 312.
The Si' species compensate the negatively-charged tetra18 J. Sauer, Proc. Int. Symp. Zeolite Catal., Siofok (Hungary), 1985,
hedral aluminium atoms of the intact framework. While it
p. 19.
was not possible to verify the existence of this low concentra19 J. Sauer and P. Hobza, Theor. Chim. Acto (Berlin), 1984,65,279;
tion of threefold coordinated framework atoms experimen1984,65, 291.
tally using X-ray34 and NMR2' spectrometry, K a ~ a n s k y ~ ~20 V. M. Mastikhin and 0. B. Lapina, React. Kinet. Catal. Lett.,
has found an TR band at 4035 cm-' for H, adsorbed on an
1979, 11, 353.
21 J. W. Akitt, J. Chem. SOC. A, 1971,18,2865.
H-Y zeolite activated at 770 K under deep-bed conditions
22 D. Freude, T. Frohlich, M. Hunger, H. Pfeifer and G. Scheler,
which he ascribed to H, adsorbed on threefold coordinated
Chem. Phys. Lett., 1983,98, 263.
and positively-charged silicon atoms of the framework
23 E. T. Olejniczak, S. Vega and R. G. Grifin, J. Chem. Phys., 1984,
according to scheme (2). In accordance with this finding we
81,4804.
assume that the water molecules which give rise to the signal
24 D. Freude, W. Oehme, H. Schmiedel and B. Staudte, J. Catal.,
at 6.5 ppm are also adsorbed on these threefold coordinated
1977,49, 123.
25 C. E. Bronnimann, R. C. Zeigler and G. 13. Maciel, J. Am. Chem.
and positively-charged silicon atoms of the zeolite frameSoc., 1988, 110, 2023.
work. This interpretation is not at variance with the results of
26 J. W. Emsley, J. Feeney and L. M. Sutciiffe, Nigh Resolution
X-ray34 and NMR2, spectrometry since the concentration of
N M R Spectroscopy, Pergamon Press, Oxford, 1966, p. 263.
these sites (2.0 k 0.5 per unit cell, cf. Table 1) amounts to
27 J . Scherzer, ACS Symp. Ser., 1984,248, 157.
< 1 % of the T-atoms which is below the limits of detection of 28 R. Bertram, U. Lohse and W. Gessner, Z . Anorg AIlg Chem.,
the methods used in ref. 34 and 22. Under this assumption
1988, 567, 145.
29 P. K. Maher, F. D. Hunter and J. Scherzer, Ado. Chem. Ser.,
the heteronuclear magnetic dipolar interaction of the water
1971,101,266.
protons is expected to be much smaller than that for the
30 M. Hunger, D. Freude, H. Pfeifer, D. Prager and W. Resadsorption on extraframework aluminium cations, in agreechetilowski, Chem. Phys. Lett., 1989, 163, 221.
ment with the experimentally observed small intensity of the
31 J. Karger, H. Pfeifer and W. Heink, Adti. Magn. Reson., 1988, 12,
MAS sidebands ( < 20%).
1.
32 H. Pfeifer, Phys. Rep., 1976, 26, 293.
33 R. C. Weast and M. J. Astle, CRC Handbook of Chemistry and
Physics, CRC Press, Boca Raton, FL, 63rd edn., 1983, p. F-183.
References
34 G. H. Kuhl, J. Phys. Chem. Solids, 1977,38, 1259.
1 A. J. Vega and Z. Luz, J. Phys. Chem., 1987,91,365.
35 V. B. Kazansky, Catal. Today, 1988,3.,367.
2 Z. Luz and A. J. Vega, J. Phys. Chem., 1987,91, 374.
3 A. J. Vega, J. Am. Chem. Soc., 1988,110,1049.
4 C. E. Bronnimann, I. S. Chuang, B. L. Hawkin and G. E. Maciel,
J . Am. Chem. Soc., 1987,109, 1562.
Paper 0/020551; Received 9th May, 1990
P\
9,-?\
-
P P