Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
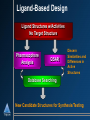
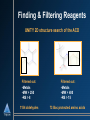
Computational Techniques in Support of Drug Discovery Jeffrey Wolbach, Ph. D. October 2, 2002 Who Is Tripos? Discovery Software & Methods Research Core Science & Technology Chemistry Products & Services Software Consulting Services Discovery Research & Process Implementation Sequential Drug Discovery Choose Disease Target Identification Target Validation Lead Identification Lead Validation Lead Optimization ADME Candidate to Clinic Many cycles of synthesis/testing to identify and optimize lead Role of molecular modeling o o o o unrealistic to jump from validated target to optimized lead useful to reduce the number of synthesis/testing cycles enables “first to file” enlarge number of targets Drug Discovery in Parallel • Choose a Disease Target Identification Target Validation Knowledge-sharing environment: genomics, HTS, chemistry, ADME, toxicology • Collect more data, on more compounds, more quickly • Apply predictive models of “developability” early Lead Identification Lead Validation • • Enhanced understanding & predictive model building Increase share of patented time on market Lead Optimization ADME Candidate to Clinic Ligand-Based Design Ligand Structures w/Activities No Target Structure Pharmacophore Analysis QSAR Discern Similarities and Differences in Active Structures Database Searching New Candidate Structures for Synthesis/Testing Pharmacophore Analysis • Assume active molecules share a binding mode o • Don’t know binding mode, so active molecules are considered flexible o o • Search for common chemical features of active molecules Search set of pre-determined conformers Allow molecules to flex during search Typical features: o o o H-Bond Donors H-bond acceptors Hydrophobic groups Pharmacophore Models • • Chemical features in 3-D space Distance constraints between chemical features QSAR • • Relates bioactivity differences to molecular structure differences Structure represented by numerical descriptors o o • Traditional (2D) QSAR 3D QSAR - CoMFA Statistical techniques relate descriptors to activity + D Activity + + ++ + ++ ++ Activity = D0 + 0.5 D1 + 0.17 D2 + ... + + D Descriptor QSAR - Traditional (2D) • Descriptors are molecular properties o logP, dipole moment, connectivity indices ... Structures + Activity pKi=5.3 pKi=3.7 pKi=2.9 Descriptors Predictive Model (QSAR Equation) logP = 1.9 m = 2.8 Estate = 7.2 pKi=A + B(logP) + C(m) + D(Estate) + ... logP = 1.7 m = 2.3 Estate = 6.7 logP = 2.1 m = 3.5 Estate = 5.5 PLS MLR . . QSAR - 3D QSAR - CoMFA • • • Comparative Molecular Field Analysis Descriptors are field strengths around molecules electrostatic, steric, H-bond .. Fields can have easy physical interpretation pKi=A + B(D1) + C(D2) + ... QSAR/CoMFA - Interpretation • High Coefficient (important) lattice points can be plotted around molecular structures 2D Database Searching O OH O O N S N O N O • 010110010010101 O Searches often performed on bit-strings o o “Fingerprints” (many types) Fingerprints display neighborhood behavior • Also includes substructure searching • Can search for similarity or dissimilarity 3D Database Searching • Query is a collection of features in 3-D space o o • Pharmacophore Lead compound / specific atomic groups Search a database of flexible, 3-D molecules o o Molecules can’t be stored in every possible conformation Allow molecules to flex to fit the query Example of Structure-Based Design 3D Database Searching • • Not restricted to ligand-based design Information about target can be included in the query o o o Can define steric hindrances Additional interaction sites Serves to filter hits from the search Identification of Novel Matrix Metalloproteinase (MMP) Inhibitors MMPs •Zinc-dependent proteases •Involved in the degradation and remodeling of the extracellular matrix They are important therapeutic targets with indications in: A fibroblast collagenase-1 complexed with a diphenylether sulphone-based hydroxamic acid •Cancer •Arthritis •Autoimmunity •Cardiovascular disease Objectives Design high affinity MMP inhibitors based on the diketopiperazine scaffold by: •Creating a virtual combinatorial library of candidate inhibitors •Using virtual screening tools to identify candidates with the highest predicted affinity •Perform R-group and binding mode analysis to guide library design Synthesis of DKP-MMP inhibitors DKP-I DKP-II 1.) Esterification of the solid support (HO-) with an amino acid 2.) Reductive alkylation of the amino acid and acylation of the resulting secondary amine 3.) Deprotection of the N-alkylated dimer followed by cyclic cleavage from the resin yielding diketopiperazine (DKP) Finding & Filtering Reagents UNITY 2D structure search of the ACD Filtered out: •Metals •MW > 250 •RB > 8 Filtered out: •Metals •MW > 400 •RB > 15 1154 aldehydes 73 Boc protected amino acids Selecting Reagents & Building the Virtual Library Selector™ Legion™ Diverse selection of amino acids (R1) and aldehydes (R2) using: Model the reaction and create virtual combinatorial library •2D Finger Prints •Atom Pairs •Hierarchical Clustering Randomly selected 14 amino acids for R3 55 amino acids x 95 aldehydes x 14 amino acids = 73,150 compounds (R1) (R2) (R3) (~75k, 8.5 MB) The CombiFlexX Protocol •Select a diverse subset of compounds using OptiSim •Dock and score the compounds in the diverse subset using FlexX •Select unique core placements using OptiSim •Hold each core placement fixed in the binding site as each R-group is independently attached, docked, and scored. •Sum the scores of the "R-cores" and subtract the score of the common core Computation times scale as the sum of the number of Rgroups rather than as the product of the number of R-groups Virtual Screening of DKP-MMP Inhibitors • ~75k compound library • MMP target structure collagenase-1 (966c.pdb) • 150 diverse compounds selected and docked • 39 non-redundant core placements based on RMSD > 1.5 Å. Virtual Screening Results •Docked 91% of the library •36 compounds/minute •331 compounds predicted to be more active than those published Consensus Scoring Results •Extracted the top 1000 library compounds based on Flex-X score •Ranked the top library compounds and published “highly actives” using CScore •10 compounds predicted by all scoring functions to be more active than “highly actives” R-Group Analysis in HiVol Frequency of R-group use among 331 active virtual compounds R1 (55 reagents) R2 (95 reagents) R3 (14 reagents) R-Group Analysis in HiVol, con’t. Frequency of R-group use among 331 active virtual compounds R1 Summary Used CombiFlexX and HiVol to: • Identify highly promising candidates • Perform R-group analysis & Binding mode analysis to guide further computational design of libraries Further Work • Diversity/similarity analysis of the published and virtual libraries • Use docking results for library design in Diverse Solutions • SAR development Binding Mode Analysis Frequency of core placement use R-Group Analysis in HiVol Frequency of R-group use among 331 active virtual compounds R1 (55 reagents) R2 (95 reagents) R3 (14 reagents)