Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Mammary gland wikipedia , lookup
Hyperthyroidism wikipedia , lookup
Neuroendocrine tumor wikipedia , lookup
Polycystic ovary syndrome wikipedia , lookup
Norepinephrine wikipedia , lookup
Triclocarban wikipedia , lookup
Growth hormone therapy wikipedia , lookup
Hormone replacement therapy (male-to-female) wikipedia , lookup
Hypothalamus wikipedia , lookup
Hypothalamic–pituitary–adrenal axis wikipedia , lookup
History of catecholamine research wikipedia , lookup
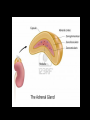
Introduction The adrenal gland is a multifunctional organ that produces the steroid hormones and neuropeptides which are essential for life. Despite the complex effects of adrenal hormones, most pathologic conditions of the adrenal gland are linked by their impact on blood pressure and electrolyte balance. In clinical practice, patients often present with states of diminished production or overproduction of one or more adrenal hormones. Hypofunction is generally treated with exogenous hormone replacement, and hyperfunction is generally treated with pharmacologic suppression or surgery. Anatomy of adrenal gland The two adrenal glands (also called the suprarenal glands) are situated in the abdomen, above the kidneys and below the diaphragm. They have a high cholesterol content giving them a yellowish color. They are contained within the same membrane as the kidney but separated from them by a fibrous layer of tissue. The right gland is tetrahedral in shape and lies lower than the left, which is semilunar in shape and usually the larger of the two. When cut in half each gland consists of an outer cortex, yellow in color and an inner medulla, which is dark red, or grey. The cortex consists of three distinct zones. They are: - Zona glomerulosa - Zona fasciculata - Zona reticularis Each zone has a characteristic histology and secretes different types of hormones. Blood supply Adrenal arterial supply is symmetric. Small arterioles branch to form a dense subcapsular plexus that drains into the sinusoidal plexus of the cortex. There is no direct supply to middle and inner zones. Venous drainage from the central vein displays laterality. After crossing the medulla, the right adrenal vein empties into the inferior vena cava, and the left adrenal vein drains into the left renal vein. Adrenal cortex The function of the cortex is to produce adrenocorticoid hormones. Mineralocorticoids Glucocorticoids Androgens )Sex steroids) Mineralocorticoids: Zona glomerulosa cells (outer 10%) synthesize mineralocorticoids, the most important hormone is aldosterone, which account for 90% of mineralocorticoids activity. It helps in maintaining the balance of water and salt in the body. Aldosterone helps in regulating the amount of sodium that is excreted into the urine. The production of aldosterone is regulated by the renin angiotensin. Whenever the blood pressure fluctuates or the balance of salt and water is disturbed. Glucocorticoids:• Zona fasciculata cells (middle 75%) synthesize glucocorticoids, such as cortisol. • The adrenal cortex releases corticosteroids that help the body to deal with stressful situations. • Cortisol is regulated by the brain's hypothalamus and the pituitary gland. First, the hypothalamus releases hormone called corticotropin-releasing hormone (CRH) that signals the pituitary gland to respond by sending out ACTH, which in turn stimulates the adrenal glands to respond by producing cortisol. Cortisol released to raise blood sugar, blood pressure levels, and strengthens the immune system. If the cortisol level is low, the person has:1. Fatigue 2. low blood pressure 3. Hypoglycemia 4. Poor immune function 5. Increased tendency to allergies and environmental sensitivity 6. Inability to deal with stress. Excess Cortisol result in:1. Diminished cellular utilization of glucose, thus increases blood sugar levels. 2. Decreases protein synthesis. 3. Increases protein breakdown that can lead to muscle wasting. 4. Causes demineralization of bone that can lead to osteoporosis. 5. Diminishes lymphocyte numbers and function. Androgens (Sex steroids):The fasciculata also generate androgen precursors such as dehydroepiandrosterone (DHEA), which is sulfated in the innermost zona reticularis to dehydroepiandrosterone sulfate (DHEAS) Functions of DHEA:1. Is a precursor that is converted to testosterone (a male hormone). 2. Is a precursor to estrogen (a female anabolic hormone). 3. Reverses immune suppression caused by excess cortisol levels, thereby improving resistance against viruses, bacteria and Candida albicans, parasites, allergies, and cancer. 4. Stimulates bone deposition and remodeling to prevent osteoporosis. 5. Increases muscle mass. 6. Decreases percentage of body fat. 7. Involved in the thyroid gland's conversion of the less active T4 to the more active T3. 8. Improves cardiovascular status by lowering total cholesterol and LDL levels, thereby lessening incidences of heart attack. 9. Accelerates recovery from any kind of acute stress (e.g., insufficient sleep, excessive exercise, mental strain, etc.). Adrenal Steroidogenesis • synthesis of the steroid hormones produced by the adrenal cortex . • All steroid hormones are derived from cholesterol, which taken up by the cell in the form of low density lipoprotein (LDL). • LDL is taken into cells via LDL receptors, and broken down into esterified cholesterol, and then free cholesterol. • A series of enzymatic steps in the mitochondria and ER of steroidogenic tissues convert cholesterol into all of the other steroid hormones and intermediates. • The first enzymatic step is the conversion of cholesterol to pregnenolone, by enzyme cytochrome P450. • This step occurs in the mitochondria, in the adrenal, ovary, and testis. • Next, pregnenolone can be converted into three different pathways, depending upon whether you want to make mineralcorticoids, glucocorticoids, or androgens. Congenital adrenal hyperplasia Congenital adrenal hyperplasia (CAH) encompasses a group of autosomal recessive disorders, each of which involves a deficiency of an enzyme involved in the synthesis of cortisol, aldosterone, or both. The clinical presentation depends on the affected enzyme, with 95% result of a 21-hydroxylase deficiency Deficiency in 21-hydroxylase cause 17-OH progesterone and androgen buildup while cortisol decreases. This deficiency inherited in severe or mild forms. The severe form, called Classical CAH, is usually detected in the newborn period or in early childhood. The milder form, called Non-classical CAH (NCAH), may cause symptoms at anytime from infancy through adulthood. NCAH is a much more common disorder than Classical CAH. When the pituitary gland senses that there is not enough cortisol present in the bloodstream, it releases a hormone ACTH to stimulates the adrenals to produce more cortisol. Patient with CAH have insufficient amounts of the enzyme 21hydroxylase, needed to convert 17-hydroxyprogesterone (17OHP) into cortisol. As a result, the pituitary gland continues to sense the need for cortisol and pumps out more ACTH. This leads to an overabundance of 17-OHP, which is converted in the adrenals into excess androgens. Lack of adequate cortisol also prevents the body from properly metabolizing sugar and responding to stress. The lack of this stress response can lead to an adrenal crisis. In addition, over 75% of all individuals with classical CAH also lack another adrenal hormone called aldosterone. When this deficiency occurs it is called “Salt-Wasting CAH” (SW-CAH). The remaining 25% of those with Classical CAH who produce sufficient aldosterone are referred to as “Simple Virilizers” (SVCAH). Hypoaldosteronism Insufficient aldosterone secretion is seen with:- Adrenal gland destruction. Chronic heparin therapy. Following unilateral adrenalectomy (transient). In G-layer enzyme deficiencies. Most hypoaldosteronism occurs in patients with mild renal insufficiency such as persons with diabetes who present with mild metabolic acidosis, high serum potassium, low urinary potassium excretion (urine K urine Na), and low reninemia. Hyperaldosteronism Patients with excess aldosterone production may develop metabolic alkalosis, HTN, and hypokalemia. Causes of HTN and unprovoked hypokalemia include: - Primary aldosteronism (low renin)—autonomous oversecretion of aldosterone - Secondary aldosteronism (elevated renin)—RAS activated aldosterone secretion - Pseudoaldosteronism (variable renin and aldosterone levels) Primary adrenal insufficiency Primary adrenal insufficiency, also known as Addison's disease, occurs when the adrenal glands cannot produce an adequate amount of hormones despite a normal or increased ACTH level. Causes of primary adrenal insufficiency:1- autoimmune adrenalitis (slow destruction of the adrenal cortex by cytotoxic lymphocytes). 2- autoimmune thyroid disease and other autoimmune endocrine deficiencies ( that develop antibodies against the steroidogenic enzyme 21-hydroxylase). 3- Acquired immunodeficiency syndrome (AIDS), in which the adrenal gland may be destroyed by a variety of opportunistic infectious agents. All causes of primary adrenal insufficiency involve the adrenal cortex as a whole, resulting in a deficiency of cortisol and aldosterone (plus adrenal androgen). Secondary and tertiary adrenal insufficiency In secondary adrenal insufficiency, an insufficient amount of ACTH is produced by the pituitary gland. In tertiary adrenal insufficiency, an insufficient amount of CRH is produced by the hypothalamus. Causes of secondary adrenal insufficiency:- Glucocorticoid therapy - Tumors - Hemorrhage - Infiltrative processes - Malignancies Low Cortisol (baseline) None to minimal cortisol stimulation Primary adrenal insufficiency High ACTH (baseline) Low aldo (or no stimulation?) Hyperpigmentation ±hyperkalemia/hyponatremia ±virilization Normal cortisol stimulation(can be blunted with chronic insufficiency) Secondary adrenal insufficiency Low ACTH (baseline) Aldo normal (stimulation? >4 ng/dL) Subtle symptoms Electrolytes OK (Aldo preserved) Differential diagnosis of low cortisol states Cushing syndrome Cushing’s syndrome result from excess glucocorticoid production or prolonged exogenous steroid use. The most common causes of Cushing’s syndrome are:• ACTH-secreting pituitary adenoma (68%). • Autonomous cortisol production from an adrenal tumor (17%, ACTH is suppressed). • Excess ectopic ACTH or CRH production (15%, usually malignant). Causes of Cushing’s Syndrome (High Cortisol) by ACTH High ACTH Cushing’s with hyperpigmentation ACTH Dependent - Primary ACTH (pituitary disease) - Ectopic ACTH - Ectopic CRF Low ACTH ACTH Independent - Adrenal adenoma - Adrenal carcinoma - Nodular adrenal hyperplasia - Exogenous glucorticoids Differentiating source of ACTH secretion ANDROGEN EXCESS In women, androgen overproduction can cause infertility, with masculinizing effects (e.g., hirsutism, acne, male pattern baldness, menstrual irregularities, and virility). In men, excess adrenal androgens can also cause infertility with feminizing effects by inhibiting pituitary gonadotropins, which effectively lowers testicular testosterone production. Diagnosis of Excess Androgen Production <10% of DHEAS and DHEA are produced by the gonads, therefore, high DHEAS and DHEA production strongly suggests adrenal hyperandrogenism, whereas elevated testosterone values are seen with either adrenal or gonadal hyperandrogenism. Plasma DHEAS, DHEA, or urine 17-ketosteroids can identify patients with adrenal causes of pathologic masculinization (females) and feminization (males). Catecholamine In response to stimulation, the medulla secretes catecholamines directly into the circulation. Medullary catecholamine products serve as first responders to stress by acting within seconds (cortisol takes 20 minutes) to promote the fight-or-flight response, which increases cardiac output and blood pressure, diverts blood toward muscle and brain, and mobilizes fuel from storage. Catecholamines are hydrophilic, circulate in low levels (50% albumin bound), have short half-lives (seconds to 2 minutes). The most abundant catecholamines are epinephrine (adrenaline) and norepinephrine (noradrenaline) which synthesized from phenylalanine and tyrosine. The ratio of NE to EPI in the serum is normally 9:1 (98% from postganglionic neurons, 2% from the medulla). Cytoplasm Phenylalanine Secretory Vesicle EPI Dopamine VMAT Tyrosine DOPA NE lipid vesicles Dopamine VMAT EPI 80% of adrenal catecholamine PNMT Cortisol Biosynthesis and storage of catecholamines PNMT :- phenylethanolamine N-methyltransferase VMAT:- vesicle monoamine transporters Catecholamine Degradation:All catecholamines are rapidly eliminated from target cells and the circulation by three mechanisms: 1. Reuptake into secretory vesicles 2. Uptake in nonneuronal cells (mostly liver) 3. Degradation Degradation relies on two enzymes:- catechol methyltransferase (COMT) (in nonneuronal tissues) - monoamine oxidase (MAO) (within neurons) Catecolamines degraded to produce metabolites (metanephrines and VMA) from free catecholamines. Metabolites and free catecholamines are eliminated by direct filtration into the urine and excreted as free NE (5%), conjugated NE (8%), metanephrines (20%), and VMA (30%). Urine EPI (50%) is converted from NE by renal, not adrenal, before excretion. Methods of analysis:• Urine catecholamines (free NE and EPI) are assayed using liquid chromatography, fluorometry, and liquid chromatography (LC) tandem mass spectrometry. • 24hr urine catecholamine and metabolite levels are more reliable and are not altered by age or gender. • Most antihypertensive drugs and many other medications interfere with accurate catecholamine measurement by fluorometric assays. Substances causing autofluorescence (e.g., tetracyclines, ephedrine, methlyldopa) can produce erroneous results when measured by fluorometric assays. Pheochromocytoma Pheochromocytomas are rare catecholamine producing tumors arising from chromaffin tissue, which causes HTN in association with nonspecific clinical symptoms that mimic anxiety. Mechanisms of catecholamine secretion by persons with pheochromocytoma remain unclear (tumors are not innervated). But characterized by:- Increased catecholamine synthesis - limited degradation capacity - limited storage for excess NE and metabolites likely cause spillover into the blood, increasing circulating free NE and/or EPI along with other active peptides that cause symptoms. Diagnosis of Pheochromocytoma • The best test for diagnosing pheochromocytoma is measurement of Catecholamine and their metabolites which include (metanephrine, normetanephrine, dopamine, and vanillylmandelic acid (VMA)) in urine or blood. • Plasma metanephrines, measured by high-performance liquid chromatography or RIA, are touted as the most specific and sensitive diagnostic test, although some investigators found that plasma metanephrines lack specificity and do not recommend it as a first-line test but reserve it for high-risk patients or patients who cannot collect an accurate 24-hour urine specimen. • Urine metanephrines may be the most sensitive urine test; it is less likely to be altered by drugs or certain foods. • 80% of pheochromocytoma patients have increased plasma chromogranin A levels. • Serum chromogranin A is is less sensitive and specific for pheochromocytoma than are direct catecholamine and metabolite measurments. • In combination, serum chromogranin A and plasma catecholamines are specific (95%) but less sensitive (88%), likely because of high dependence on renal function. Adrenal Incidentaloma Asymptomatic adrenal tumor that is discovered on an imaging test (CAT scan, MRI, etc) which was ordered to evaluate a problem that is unrelated to adrenal disease. Many adrenal masses, typically greater than 1 cm in diameter, are found incidentally. Autopsy studies report a frequency of adrenal adenomas at about 6%, and the prevalence increases with age. Although most of these lesions are nonfunctioning and benign, all should be assessed for malignancy or hypersecretion.