Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
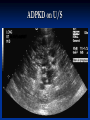
Renal Cystic Disease ADPKD ARPKD Tuberous Sclerosis Medullary Sponge Kidney Von-Hippel Lindau Disease Medullary Cystic Disease Acquired Cystic Kidney Disease Multicystic Renal Dysplasia Mark D. Purcell DO Nephrology/Internal Medicine Carolina Nephrology, PA 203 Mills Avenue Greenville, SC 29605 (864) 271-1844 [email protected] Case Presentation 54 y.o. Caucasian male C.C.: “Concerned about developing cysts in kidneys. 3 of 4 of my siblings have it!” PMH: Negative PSH: Negative Meds: Rarely OTC NSAIDs Allergies: None Case Presentation ROS: Negative Physical exam: VSS No abnormal findings on exam Labs: Cr=1.1 Hgb=14.2 UA: Neg for protein or blood, RBC=2-4, WBC=0 Questions? “Do I have Cystic kidneys?” “If not, what are my chances of developing Cystic kidneys in the future?” “If I have it, what is the treatment?” “I heard some could develop a stroke, right?” “Could my children get tested to see whether they have it?” “What are my chances of needing dialysis?” Differential Diagnosis Simple renal cysts The most common renal masses Solitary, multiple and bilateral Occur most often over age 50 Often seen on screening ultrasound exam Range in size from less than 1cm to 10cm Usually do not compromise renal function Simple renal cysts Usually one epithelial layer without renal elements Differential diagnosis : rule out more serious disorders Polycystic kidney disease: family history, bilateral, gross hematuria, infection Loculated cystic disease: unilateral Malignancy: thickened irregular walls, septae, enhance with contrast, multilocular mass Simple renal cysts Treatment: Usually none required Pain: rare, Acetaminophen, NSAIDs short course if renal function preserved Rare if ever aspiration and surgery Infection: Rare Bosniak Classification Based on morphologic and enhancement characteristics of cysts by CT scanning Used to help diagnose and manage cystic lesions in the kidney Categories from I to IV based on increasing likelihood of malignancy Simple Cyst Simple cyst Autosomal Dominant PKD (ADPKD) Autosomal Dominant PKD (ADPKD) Presentation of ADPKD Usual presentations: Screening with positive family history Flank pain, or Kidney Disease On Ultrasound or CT, large kidneys with multiple bilateral cysts are seen Cysts may also be seen in the liver, pancreas, and spleen Families with well known mutations in PKD genes Clinical Manifestations Extra-renal manifestations Cerebral Aneurysms Hepatic cysts (most common extra-renal manifestation) 4% young adults up to 10% in older patients Role of screening uncertain (MRI vs CT angiogram) High risk patients: family history, require anticoagulation, pre-op, high risk occupation, symptomatic patients Occur later than renal cysts, increase with age Cardiac valve disease: 25-35% of patients Colonic diverticula: seen more in patients on dialysis Abdominal wall inguinal hernias U/S and Renal Cysts J Am Soc Nephrol 13(9):2384-98, 2002 Acquired Cysts vs. ADPKD Patients with advanced renal failure may present with acquired multiple renal cysts on ultrasonography or CT These kidneys, however, have a normal or reduced size and the renal contour is smooth Many patients have known pre-existent renal disease There is also no family history of PKD ADPKD: U/S Diagnostic Criteria ADPKD on U/S ADPKD on CT Epidemiology and Genetics ADPKD is by far the most commonly inherited kidney disease Prevalence ranges from 1-2.5 % Characterized by the development of multiple renal cysts Associated with extra-renal manifestations Caused by mutations identified in at least three genes (PKD1, PKD2, PKD3) N Engl J Med 329(5):332-42, 1993 Genetics J Am Soc Nephrol 13(9):2384-98, 2002 Genetics PKD1 is an extremely large and complex gene (46 exons) 14 kb mRNA Polycystin 1 is over 4000 amino acids PKD1 gene is adjacent to one of the genes for tuberous sclerosis (TSC2), a different disorder that is also associated with cyst (angiomyolipoma) formation in the kidneys J Am Soc Nephrol 13(9):2384-98, 2002 Genetics PKD2 is a much smaller gene Encodes a much smaller mRNA Polycystin 2 is a little smaller than 1000 amino acids J Am Soc Nephrol 13(9):2384-98, 2002 Polycystin 1 and 2 Polycystin-1 Polycystin-1 is localized in renal tubular epithelia, hepatic and pancreatic ducts, and all sites of cyst formation in ADPKD It is an integral membrane protein, found primarily in plasma membranes It is over-expressed in most but not all cysts in kidneys from patients with ADPKD J Am Soc Nephrol 13(9):2384-98, 2002 ADPKD pathophysiology PKD 1 gene codes a large and complex protein (polycystin-1) Polycystin 1 acts as a receptor that binds to and activates polycystin 2 leads to rapid flux of Ca++ through cell membrane cation channels G- protein binding site is activated Normal tubular morphogenesis and growth Mutations in the polycystin gene with promote abnormal growth of cysts A second “hit” or somatic mutation of the allele inherited from the normal parent is required for cyst formation Double-hit and Cyst Formation Kidney Int 67(4):1234-47, 2005 Cyst Formation Protein-protein, cell-cell, and cell-matrix interaction defects Impaired tubulogenesis in cells derived from ADPKD cysts in vitro Abnormalities in renal cilia due to polycystin mutations contribute to renal cyst formation. Certain genetic defects in cilia formation and function are related to cyst development J Am Soc Nephrol 13(9):2384-98, 2002 Cyst Formation Fluid secretion in the cysts may be mediated in part by the cystic fibrosis transmembrane conductance regulator (TCR) Individuals with both ADPKD and cystic fibrosis may have less severe cystic disease of the kidney and liver compared to family members with ADPKD but without cystic fibrosis Renal Manifestations Hematuria Can be gross or microscopic Isosthenuria Proteinuria Nephrolithiasis Pain Cancer Anemia Hematuria May be macroscopic Incidence 35-50% Macroscopic hematuria associated with worse outcome Etiology: UTI Nephrolithiasis Strenuous activity RCC ?Cyst rupture Am J Kidney Dis 20(2):140-3, 1992 Isosthenuria Many have a mild concentrating defect 177 adult patients with normal kidney function, all had a modest impairment in concentrating ability in response to overnight water deprivation, which worsened with age in parallel to the decline in concentrating ability of unaffected family members Kidney Int. 35(2):675-80, 1989 The concentrating defect may not be clinically evident except if a history of polydypsia or polyuria is elicited The underlying cause is not known, but disruption of tubular architecture, defect in principal cell function, or early tubulointerstitial disease are postulated factors A central cause has been excluded since vasopressin levels are elevated in this disorder Kidney Int. 68(5):2405-18, 2005 Proteinuria Not very common Usually <1 g/d Parallel to renal function Nephrotic syndrome usually means superimposed GN N Engl J Med 329(5):332-42, 1993 Nephrolithiasis Up to 20% of patients >50% uric acid Rest is mostly calcium oxalate ESWL successful with small stones Risks Low urine volume low urinary citrate less often hyperuricosuria and hypercalciuria Am J Kidney Dis 22(4):513-9, 1993 Pain Abdominal and flank pain Infection Nephrolithiasis Cyst rupture/hemorrhage Renal Cell Carcinoma Mayo Clinic and Foundation, Rochester 25 patients No male predominance was observed (12 men, 13 women) The age of presentation was earlier than that seen in the general population (45 versus 61 yr) Fever, night sweats, and weight loss were prominent at presentation Fever is a more common presenting symptom of renal cell carcinoma in ADPKD (32% vs 7%) 20% of the patients had metastatic disease at presentation Even with computed tomography and magnetic resonance, the diagnosis was difficult and often delayed WBC scan can wrongly suggest a cyst infection More often concurrently bilateral (12 versus 1 to 5%), multicentric (28 versus 6%), and sarcomatoid in type (33 versus 1 to 5%) Am Soc Nephrol 1994 Mar;4(9):1661-9 Urinary Tract Infection 30-50% lifetime incidence Similar to general population (organisms, antibiogram, presentation and treatment) Infected cyst present with a new area of discrete tenderness (pyelonephritis is associated with diffuse flank pain) For infected cysts, fluoroquinolone orally for a minimum of 4-6 weeks Am J Kidney Dis 10(2):81-8, 1987 Nephrol Dial Transplant 13:2455, 1998 Am J Kidney Dis 2006; 47:e9 Clinical findings and course Patients may present with hypertension, hematuria, flank pain, calculi, or urinary tract infection Hypertension may present with normal renal function. Renal function starts to decline usually by the fourth decade. (GFR decreases 4-6 mL/min/year) Gradual progression to End Stage Renal Disease Pain from cysts and enlarged kidneys is a common manifestation as disease progresses Clinical findings and course Risk for progression to ESRD Genotype PKD1 vs PKD2 Hypertension Early onset proteinuria and hematuria Males Increasing kidney size and rate of growth Biggest risk factor for progression to end stage renal disease (CRISP trial) Left ventricular mass index Proteinuria alone Family members who progress to ESRD Mortality National registry data: similar to other patients with end-stage renal disease and most are due to cardiac causes One report examined the cause of death in 129 pts Cardiac 36% Infection 24% Neurologic 12% (rupture ICA and bleed) Cancer 0% Pregnancy Fertility rate similar to unaffected family members Hypertensive women at increase risk of fetal loss Increase risk of ectopic pregnancy J Am Soc Nephrol 5(12):2048-56, 1995 Treatment 1. Inhibit fluid secretion to slow cyst growth: Vasopressin V2 receptor antagonists (animals) J Am Soc Nephrol 16:846, 2005 blocking sodium channels with the potassium-sparing diuretic amiloride (animals) Kidney Int 35:1379, 1989 ? Caffeine restriction (In vitro) J Am Soc Nephrol 13:2723, 2002 2. Inhibition of cell proliferation No clinical studies are available mTOR inhibitors reduces cyst formation and growth in animal models Treatment 3. Control of hypertension: renoprotection and IC bleeding Nephrol Dial Transplant 18:2314, 2003, Journal of the Renin-Angiotensin-Aldosterone System. 7(3):139-45, 2006 4. Cyst drainage: pain control, not for renal function J Am Soc Nephrol 2(7):1219-26, 1992 5. Metabolic acidosis to prevent bone disease and muscle wasting 6. Protein restriction: no benefit 7. Transarterial embolization/ Ethanol ablation Treatment No specific treatment proven to delay progression of ADPKD Rigorous blood pressure control may slow progression of ESRD and decrease risk of cardiovascular mortality If no contraindication, ACE-inhibitor should be first line therapy Statin therapy to reduce risk of cardiovascular disease Vasopressin receptor antagonists with some in vitro evidence Increased fluid intake, suppress ADH, inhibit cyst growth Inc fluids esp with stones Hemodialysis favored over peritoneal dialysis Transplantation Post-transplant erythrocytosis Diverticulitis with perforation Symptomatic aneurysms Often require unilateral or bilateral nephrectomy Avoid recurrent urinary infections Need a place to put the graft Increased patient satisfaction No benefit to survival ADPKD and ICA Screening Screening ADPKD, Mortality and ESRD Progression to ESRD, why? Compression of adjacent normal parenchyma (?) But no one knows for sure. This is a hypothesis Histologic examination of patients with early and end-stage renal failure suggests that progressive renal failure in ADPKD correlates with the development of vascular sclerosis and interstitial fibrosis Kidney Int 42(5):1259-65, 1992 Negative family history Diagnosed in up to 25% of cases Milder forms of the disease may have gone undetected in previous family members 5 % may be a new mutation No definitive diagnostic imaging criteria 10 or more cysts in each kidney Autosomal recessive PKD Incidence 1:10,000 – 1:40,000 Always associated with hepatic involvement More severe cases seen on ultrasound in utero Less severe seen with enlarging kidneys after birth Congenital hepatic fibrosis Patients alive after 1st month have 80% chance of survival to 15 and beyond Genetic defect on chromosome 6 – PKHD1 gene encodes protein fibrocystin Found in cortical and medullary collecting ducts Epithelial cells of bile ducts Autosomal recessive PKD Autosomal recessive PKD Autosomal recessive PKD Autosomal recessive PKD Autosomal recessive PKD Diagnosis and Outcomes Diagnosis usually made by ultrasound Kidneys diffusely hyperechogenic with multiple small cysts Dilated water filled collecting ducts on MR Adults may have slowly progressive kidney dysfunction Systemic hypertension develops in 75% No good screening for the gene currently Medullary sponge kidney Malformation of the terminal collecting ducts in the renal pyramids Formation of small and large medullary cysts, do not involve the cortex Usually bilateral but can be unilateral Benign disease with complications of nephrolithiasis and urinary tract infections Underlying defect is not known Often discovered by incidental radiology study such as intravenous pyelogram Flank pain, hematuria, UTIs related to stones Medullary sponge kidney Cystic dilations lead to a “brushlike” appearance on IVP. Stones may appear in clusters at the affected renal calyces Medullary sponge kidney Prognosis Impaired urinary calcium excretion leads to stones Long term prognosis excellent Rarely advances to End Stage Renal Disease Tuberous sclerosis Autosomal Dominant genetic disorder 1 in 5,000-10,000 births Only one-third of cases are familial Neurocutaneous disorder involving many organ systems Clinical diagnosis made by disease findings Two separate gene mutations TSC1 chromosome 9q34, encodes hamartin protein, expressed in normal tissues TSC2 chromosome 16p13, encodes tuberin protein, participates in normal brain development The two proteins form a complex that is thought to function as a negative regulator of the cell cycle Interaction with polycystin 1 and tuberin Clinical features: Triad seizures, mental retardation, and facial angiofibromas occur in 50% of patients Tuberous sclerosis complex Major criteria Autosomal dominant Renal angiomyolipoma Facial angiofibromas, forehead plaques Periungual fibromas Three or more hypomelanotic macules Shagreen patch Multiples retinal nodular hamartomas Cortical tubers, giant cell atrocytomas Cardiac rhabdomyoma lymphangioleiomyomatosis Minor criteria Multiple renal cysts Nonrenal hamartoma Hamartomatous rectal polyps Retinal achromic patch Cerebral white matter radial migration tracts Bone cysts Gingival fibromas Confetti skin lesions Multiple enamel pits Facial angiofibromas Ash leaf spot Fibrous plaque Angiomyolipoma Von Hippel-Lindau Disease Autosomal dominant syndrome 1 in 36,000 newborns Syndrome of benign and malignant tumors Presents in childhood, adolescence or adults Mean presentation age 26 VHL gene mutation Type I Type II: High risk for pheochromocytoma Von Hippel-Lindau disease Renal cysts Retinal hemangiomas – Most common Clear cell carcinomas of the kidney Cerebellar and spinal hemangioblastomas Pheochromocytoma Endocrine pancreatic tumors Epididymal cystadenomas Treatment Genetic counseling Early diagnosis of more invasive tumors Hemangioblastoma Medullary cystic disease Rare autosomal dominant cystic disease 30-50 new cases in U.S. each year Renal cysts at the corticomedullary junction Small to normal size kidneys Hyperuricemia and gout Grouped with other hyperuricemic diseases Nephronophthisis/MCKD complex Diagnosis and Treatment Strong family history Preponderance of gout Bland urinary sediment, no protein US- may show medullary cysts Slow progressive chronic kidney disease Allopurinol for gout Transplant is best option: disease does not recur Aquired cystic Disease Associated with hemo/peritoneal dialysis Multiple small bilateral cysts Easily distinguished from PCKD No family history Kidneys small to normal size Incidence rises with time on dialysis Pathogenesis not completely understood Nephron loss leads to hypertrophy of normal nephrons, activates proto-oncogenes, growth factors Over time tubular hyperplasia and cyst formation Dialysis associated cystic disease Most patients assymptomatic Risk of renal cell carcinoma 47% over 10 years Multicystic Renal Dysplasia Abnormal differentiation of renal parenchyma Often unilateral, opposite kidney undergoes unilateral hyperplasia Abnormal differentiation of metanephric parenchyma One of the most common forms of congenital cystic renal diseases One of the most common causes of abdominal mass in newborns Questions Mark D. Purcell, DO e-mail: [email protected]