Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
United Kingdom National DNA Database wikipedia , lookup
Therapeutic gene modulation wikipedia , lookup
Protein moonlighting wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Point mutation wikipedia , lookup
Multiple sequence alignment wikipedia , lookup
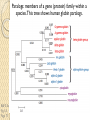
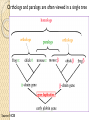
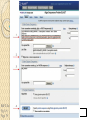
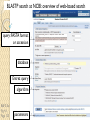
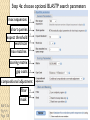
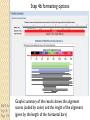
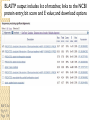
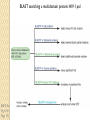
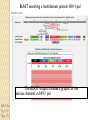
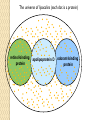
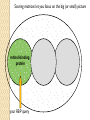
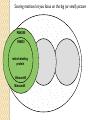
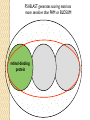
BLAST et BLAST avancé J.S. Bernardes/H. Richard Matériel : Bioinformatics and Functional Genomics Jonathan Pevsner, Wiley-Blackwell ed. Outline Introduction Definition of Orthology and Paralogy A myoglobin example BLAST search steps Step 1: Specifying Sequence of interest Step 2: Selecting BLAST Program Step 3: Selecting a Database Step 4: Selecting Search Parameters and Formatting Parameters BLAST algorithm uses local alignment search strategy BLAST algorithm parts: list, scan, extend BLAST algorithm: local alignment search statistics and E value Making sense of raw scores with bit scores BLAST algorithm: Relation Between E and p values BLAST search strategies General concepts; principles of BLAST searching How to evaluate the significance of results How to handle too many or few results BLAST searching with multidomain protein: HIV-1 Pol Using BLAST for gene discovery: Find-a-Gene Finding distantly related proteins: PSI-BLAST Learning objectives • Definition of homology, similarity, conservation • Difference between orthologues and paralogues • perform BLAST searches at the NCBI website; • understand how to vary optional BLAST search parameters; • explain the three phases of a BLAST search (compile, scan/extend, trace‐back); • define the mathematical relationship between expect values and scores; and • outline strategies for BLAST searching. Definitions: identity, similarity, conservation Homology Similarity attributed to descent from a common ancestor. Identity The extent to which two (nucleotide or amino acid) sequences are invariant. Similarity The extent to which nucleotide or protein sequences are related. It is based upon identity plus conservation. Conservation B&FG 3e Page 70 Changes at a specific position of an amino acid or (less commonly, DNA) sequence that preserve the physicochemical properties of the original residue. Globin homologs myoglobin hemoglobin B&FG 3e Fig. 3.1 Page 71 beta globin beta globin and myoglobin (aligned) Definitions: two types of homology Orthologs Homologous sequences in different species that arose from a common ancestral gene during speciation; may or may not be responsible for a similar function. Paralogs Homologous sequences within a single species that arose by gene duplication. B&FG 3e Fig. 2-3 Page 22 Myoglobin proteins: examples of orthologs B&FG 3e Fig. 3.2 Page 72 You can view the sequences at http://bioinfbook.org Paralogs: members of a gene (protein) family within a species. This tree shows human globin paralogs. B&FG 3e Fig. 3.3 Page 73 Orthologs and paralogs are often viewed in a single tree Source: NCBI Find BLAST from the home page of NCBI and select protein BLAST… Choose align two or more sequences… Enter the two sequences (as accession numbers or in the fasta format) and click BLAST. Optionally select “Algorithm parameters” and note the matrix option. sequence B&FG 3e Fig. 3-4 Page 74 Year Pairwise alignment of human beta globin (the “query”) and myoglobin (the “subject”) B&FG 3e Fig. 3-5 Page 75 We’ll examine the highlighted green region of the alignment in more detail. How raw scores are calculated: an example B&FG 3e Fig. 3-5 Page 75 For a set of aligned residues we assign scores based on matches, mismatches, gap open penalties, and gap extension penalties. These scores add up to the total raw score. Why use BLAST? BLAST searching is fundamental to understanding the relatedness of any favorite query sequence to other known proteins or DNA sequences. Applications include • identifying orthologs and paralogs • discovering new genes or proteins • discovering variants of genes or proteins • investigating expressed sequence tags (ESTs) • exploring protein structure and function BLASTP search at NCBI: overview of web-based search query: FASTA format or accession database Entrez query algorithm B&FG 3e Fig. 4-1 Page 123 parameters Outline Introduction Definition of Orthology and Paralogy A myoglobin example BLAST search steps Step 1: Specifying Sequence of interest Step 2: Selecting BLAST Program Step 3: Selecting a Database Step 4: Selecting Search Parameters and Formatting Parameters BLAST algorithm uses local alignment search strategy BLAST algorithm parts: list, scan, extend BLAST algorithm: local alignment search statistics and E value Making sense of raw scores with bit scores BLAST algorithm: Relation Between E and p values BLAST search strategies General concepts; principles of BLAST searching How to evaluate the significance of results How to handle too many or few results BLAST searching with multidomain protein: HIV-1 Pol Using BLAST for gene discovery: Find-a-Gene Finding distantly related proteins: PSI-BLAST Step 1: Choose your sequence Sequence can be input in FASTA format or as accession number BLAST step 2: choose program B&FG 3e Fig. 4-2 Page 124 Step 2. Choose the BLAST program Program Input Database 1 blastn DNA DNA 1 blastp protein protein 6 blastx DNA protein 6 tblastn protein DNA 36 tblastx DNA DNA Step 2 (choosing the BLAST program): DNA can be translated into six reading frames DNA 3 forward, 3 reverse frames protein B&FG 3e Fig. 4-3 Page 125 This image is from the NCBI Nucleotide entry for HBB Step 3: choose a database to search (protein databases) B&FG 3e Table 4-1 Page 126 Step 3: choose a database to search (nucleotide) B&FG 3e Table 4-2 Page 127 Step 4: optional parameters You can... • choose the organism to search • turn filtering on/off • change the substitution matrix • change the expect (e) value • change the word size • change the output format Example: BLASTP human insulin (NP_000198) against a C. elegans RefSeq database. Varying some parameters (filtering, compositional adjustments) can greatly affect the alignment itself. Step 4a: choose optional BLASTP search parameters max sequences short queries expect threshold word size max matches scoring matrix gap costs compositional adjustment filter mask B&FG 3e Fig. 4-4 Page 128 Step 4a: compositional adjustment influences score, expect value search results expect = 0.05 Default: conditional compositional score matrix adjustment expect = 0.09 no adjustment expect = 1e-04 B&FG 3e Fig. 4-5 Page 129 composition-based statistics Step 4b: formatting options The top of the BLAST output summarizes the query, database, and BLAST algorithm. Click to access a summary of the search parameters or a taxonomic report. B&FG 3e Fig. 4-6 Page 132 Step 4b: formatting options (you can view search parameters) Expect value BLOSUM62 matrix Threshold value T Size of database B&FG 3e Fig. 4-7 Page 133 Step 4b: formatting options B&FG 3e Fig. 4-8 Page 134 Graphic summary of the results shows the alignment scores (coded by color) and the length of the alignment (given by the length of the horizontal bars) BLASTP output includes list of matches; links to the NCBI protein entry; bit score and E value; and download options B&FG 3e Fig. 4-9 Page 134 BLAST output can be formatted to display multiple alignment B&FG 3e Fig. 4-10 Page 135 For BLASTN, CDS output displays amino acids above DNA sequence of query and subject B&FG 3e Fig. 4-11 Page 136 Outline Introduction Definition of Orthology and Paralogy A myoglobin example BLAST search steps Step 1: Specifying Sequence of interest Step 2: Selecting BLAST Program Step 3: Selecting a Database Step 4: Selecting Search Parameters and Formatting Parameters BLAST algorithm uses local alignment search strategy BLAST algorithm parts: list, scan, extend BLAST algorithm: local alignment search statistics and E value Making sense of raw scores with bit scores BLAST algorithm: Relation Between E and p values BLAST search strategies General concepts; principles of BLAST searching How to evaluate the significance of results How to handle too many or few results BLAST searching with multidomain protein: HIV-1 Pol Using BLAST for gene discovery: Find-a-Gene Perspective How a BLAST search works “The central idea of the BLAST algorithm is to confine attention to segment pairs that contain a word pair of length w with a score of at least T.” Altschul et al. (1990) How the original BLAST algorithm works: three phases Phase 1: compile a list of word pairs (w=3) above threshold T Example: for a human RBP query …FSGTWYA… (query word is in green) A list of words (w=3) is: FSG SGT GTW TWY WYA YSG TGT ATW SWY WFA FTG SVT GSW TWF WYS ... Phase 1: compile a list of words (w=3) neighborhood word hits > threshold (T=11) GTW GSW ATW NTW GTY GNW GAW neighborhood word hits < below threshold 6,5,11 6,1,11 0,5,11 0,5,11 6,5,2 22 18 16 16 13 10 9 Fig. 4.11 page 116 B&FG 3e Fig. 4-12 Page 139 Phase 2: scan the database for matches and extend B&FG 3e Fig. 4-12 Page 139 Phase 3: Traceback to generate gapped alignment B&FG 3e Fig. 4-12 Page 139 How a BLAST search works: threshold You can locally install BLAST and modify the threshold parameter. The default value for BLASTP is 11. To change it, enter “-f 16” or “-f 5” in the advanced options of BLAST+. Effect of changing the threshold T: Lower T yields more database hits (black line) and extensions (red) B&FG 3e Fig. 4-13 Page 140 For BLASTN, the word size is typically 7, 11, or 15 (EXACT match). Changing word size is like changing threshold of proteins. w=15 gives fewer matches and is faster than w=11 or w=7. For megaBLAST (see below), the word size is 28 and can be adjusted to 64. What will this do? MegaBLAST is VERY fast for finding closely related DNA sequences! How to interpret a BLAST search: expect value It is important to assess the statistical significance of search results. For global alignments, the statistics are poorly understood. For local alignments (including BLAST search results), the statistics are well understood. The scores follow an extreme value distribution (EVD) rather than a normal distribution. Normal distribution 0.40 0.35 probability 0.30 0.25 0.20 normal distribution 0.15 0.10 0.05 0 -5 -4 -3 -2 -1 0 x 1 2 3 4 Normal distribution (solid line) compared to extreme value distribution (dashed line): note EVD skewing to the right B&FG 3e Fig. 4-14 Page 141 How to interpret a BLAST search: expect value The expect value E is the number of alignments with scores greater than or equal to score S that are expected to occur by chance in a database search. An E value is related to a probability value p. The key equation describing an E value is: E = Kmn e-lS E = Kmn e-lS This equation is derived from a description of the extreme value distribution S = the score E = the expect value = the number of highscoring segment pairs (HSPs) expected to occur with a score of at least S m, n = the length of two sequences l, K = Karlin Altschul statistics Some properties of the equation E = Kmn e-lS • The value of E decreases exponentially with increasing S (higher S values correspond to better alignments).Very high scores correspond to very low E values. •The E value for aligning a pair of random sequences must be negative! Otherwise, long random alignments would acquire great scores • Parameter K describes the search space (database). • For E=1, one match with a similar score is expected to occur by chance. For a very much larger or smaller database, you would expect E to vary accordingly From raw scores to bit scores • There are two kinds of scores: raw scores (calculated from a substitution matrix) and bit scores (normalized scores) • Bit scores are comparable between different searches because they are normalized to account for the use of different scoring matrices and different database sizes S’ = bit score = (lS - lnK) / ln2 The E value corresponding to a given bit score is: E = mn 2 -S’ B&FG 3e Page 143 Bit scores allow you to compare results between different database searches, even using different scoring matrices. How to interpret BLAST: E values and p values The expect value E is the number of alignments with scores greater than or equal to score S that are expected to occur by chance in a database search. A p value is a different way of representing the significance of an alignment. p = 1 - e-E B&FG 3e Page 143 How to interpret BLAST: E values and p values E values of about 1 to 10 are far easier to interpret than corresponding p values. Very small E values are very similar to p values. __E 10 5 2 1 0.1 0.05 0.001 0.0001 ____p 0.99995460 0.99326205 0.86466472 0.63212056 0.09516258 (about 0.1) 0.04877058 (about 0.05) 0.00099950 (about 0.001) 0.00010000 E values are comparable to p values, and are designed to be more convenient to interpret. Outline Introduction Definition of Orthology and Paralogy A myoglobin example BLAST search steps Step 1: Specifying Sequence of interest Step 2: Selecting BLAST Program Step 3: Selecting a Database Step 4: Selecting Search Parameters and Formatting Parameters BLAST algorithm uses local alignment search strategy BLAST algorithm parts: list, scan, extend BLAST algorithm: local alignment search statistics and E value Making sense of raw scores with bit scores BLAST algorithm: Relation Between E and p values BLAST search strategies General concepts; principles of BLAST searching How to evaluate the significance of results How to handle too many or few results BLAST searching with multidomain protein: HIV-1 Pol Using BLAST for gene discovery: Find-a-Gene Finding distantly related proteins: PSI-BLAST Overview of BLAST search strategies B&FG 3e Fig. 4-15 Page 145 BLASTP search: human RBP4 query, human RefSeq database Results include matches (such as CG8) with high E values and limited identity to the query B&FG 3e Fig. 4-16 Page 147 “Recipricol” BLASTP search with CG8 as query includes RBP4 and other lipocalins B&FG 3e Fig. 4-17 Page 149 This confirms that the finding of CG8 using RBP4 as a query was a true positive Pairwise alignment of CG8 with non-homologous proteins B&FG 3e Fig. 4-17 Page 149 • • • • Query and subject are very different lengths E values are not significant Matches lack GXW motif Subjects are not annotated as lipocalins BLAST searching a multidomain protein: HIV-1 pol B&FG 3e Fig. 4-18 Page 151 BLAST searching a multidomain protein: HIV-1 pol The BLAST output includes a graphic of the various domains in HIV-1 pol B&FG 3e Fig. 4-19 Page 153 BLAST searching a multidomain protein: HIV-1 pol B&FG 3e Fig. 4-19 Page 153 This output shows identical residues as a dot (.). Note that the column positions that contain an arginine (R) can sometimes also contain a lysine (K) or glutamine (Q) in a position-specific pattern. This is a preview of the concept of position-specific scoring matrices (Chapter 5). Taxonomy report for a BLAST searching HIV-1 pol Most of the matches are to viruses, but there are also matches to rabbit, fungal, pig, and insect sequences. B&FG 3e Fig. 4-20 Page 154 BLASTP searching HIV-1 pol against bacterial proteins bacterial matches to HIV-1 retropepsin, reverse transcriptase domains bacterial matches to HIV-1 ribonuclease H domain B&FG 3e Fig. 4-21 Page 155 bacterial matches to HIV-1 integrase core domain BLAST searching HIV-1 pol against human sequences Question: are there human homologs of HIV-1 pol protein? Query: HIV-1 Pol Program: BLASTP Database: human nr (nonredundant) Matches: many human proteins share significant identity. B&FG 3e Fig. 4-22 Page 156 Question: are there human RNA transcripts corresponding to HIV-1 pol? Query: HIV-1 Pol Program: TBLASTN Database: human ESTs Matches: many human genes are actively transcribed to generate transcripts homologous to HIV-1 pol. Outline Introduction BLAST search steps Step 1: Specifying sequence of interest Step 2: Selecting BLAST program Step 3: Selecting a database Step 4: Selecting search parameters and formatting parameters Stand-alone BLAST BLAST algorithm uses local alignment search strategy BLAST algorithm parts: list, scan, extend BLAST algorithm: local alignment search statistics and E value Making sense of raw scores with bit scores BLAST algorithm: relation between E and p values BLAST search strategies General concepts; principles of BLAST searching How to evaluate the significance of results How to handle too many or too few results BLAST searching with multidomain protein: HIV-1 Pol “Find-a-gene project” to practice BLAST Start with the sequence of a known protein TBLASTN Inspect the output BLASTX nr or BLASTP nr B&FG 3e Fig. 4-23 Page 157 “Find-a-gene project” example: novel globin Query: NP_000509 Program: TBLASTN Database: EST (nematodes) Match: novel globin B&FG 3e Fig. 4-24 Page 158 Confirmation Query: nematode EST Program: BLASTX Best match: a globin, but not a previously annotated globin “Find-a-gene project” • The find-a-gene project is meant to be a very focused, specific project to help you understand how to use various BLAST tools (e.g. TBLASTN, BLASTX, BLASTP) and various databases. • You can start with (almost) any protein, from the organism of your choice, and discover a “novel” gene in another organism that is homologous but has never been annotated before as related to your query. Therefore you are discovering a new gene. • You can take your new gene/protein, name it, then search it against databases to confirm it has not been described before. • You can further perform multiple sequence alignment (Chapter 6), phylogeny (Chapter 7), and predict its protein structure (Chapter 13) and its function (Chapter 14). Outline Introduction BLAST search steps Step 1: Specifying sequence of interest Step 2: Selecting BLAST program Step 3: Selecting a database Step 4: Selecting search parameters and formatting parameters Stand-alone BLAST BLAST algorithm uses local alignment search strategy BLAST algorithm parts: list, scan, extend BLAST algorithm: local alignment search statistics and E value Making sense of raw scores with bit scores BLAST algorithm: relation between E and p values BLAST search strategies General concepts; principles of BLAST searching How to evaluate the significance of results How to handle too many or too few results BLAST searching with multidomain protein: HIV-1 Pol Three problems standard BLAST cannot solve [1] Use human beta globin as a query against human RefSeq proteins, and BLASTP does not “find” human myoglobin. This is because the two proteins are too distantly related. PSI-BLAST at NCBI as well as hidden Markov models easily solve this problem. [2] How can we search using 10,000 base pairs as a query, or even millions of base pairs? Many BLAST-like tools for genomic DNA are available such as PatternHunter, Megablast, BLAT, and LASTZ. [3] How can we align tens of millions of short reads to a reference genome? Position specific iterated BLAST: PSI-BLAST The purpose of PSI-BLAST is to look deeper into the database for matches to your query protein sequence by employing a scoring matrix that is customized to your query. PSI-BLAST is performed in five steps [1] Select a query and search it against a protein database B&FG 3e Page 172 PSI-BLAST is performed in five steps [1] Select a query and search it against a protein database [2] PSI-BLAST constructs a multiple sequence alignment then creates a “profile” or specialized position-specific scoring matrix (PSSM) B&FG 3e Page 172 Inspect the BLASTP output to identify empirical “rules” regarding amino acids tolerated at each position R,I,K C D,E,T K,R,T N,L,Y,G 1 M 2 K 3 W 4 V 5 W 6 A 7 L 8 L 9 L 10 L 11 A 12 A 13 W 14 A 15 A 16 A ... 37 S 38 G 39 T 40 W 41 Y 42 A A -1 -1 -3 0 -3 5 -2 -1 -1 -2 5 5 -2 3 2 4 R N D C Q E G H -2 -2 -3 -2 -1 -2 -3 -2 1 0 1 -4 2 4 -2 0 -3 -4 -5 -3 -2 -3 -3 20 -3 -3 -3 -4 -1 -3 -3 -4 -4 -3 -4 -5 -3 -2 -3 -3 -3 -2 -2 -2 -1 -1 -1 0 -2 -2 -4 -4 -1 -2 -3 -4 -3 -3 -3 -4 -1 -3 -3 -4 -3 -3 -4 -4 -1 -2 -3 -4 -3 -2 -4 -4 -1 -2 -3 -4 -3 -2 -2 -2 -1 -1 -1 0 -2 -2 -2 -2 -1 -1 -1 0 -2 all the amino acids -3 -4 -4 -2 -2 -3 -4 -3 from -2 -1 position -2 -1 -1 1 -2to 4the -2 -1 0 -1 -2 2 0 2 -1 end of your PSI-BLAST -2 -1 -2 -1 -1 -1 3 -2 2 0 0 -3 -2 4 -1 -3 -1 -3 -2 -2 query protein 0 -1 0 -4 -2 -2 -1 -2 -1 -5 -3 -2 -1 -3 -1 -3 -3 -1 0 -2 -1 -2 -2 -1 I L K M F 1 2 -2 6 0 -3 -3 3 -2 -4 -3 -2 -3 -2 1 amino acids 3 1 -3 1 -1 -3 -2 -3 -2 1 -2 -2 -1 -1 -3 2 4 -3 2 0 2 2 -3 1 3 2 4 -3 2 0 2 4 -3 2 0 -2 -2 -1 -1 -3 -2 -2 -1 -1 -3 1 4 -3 2 1 -2 -2 -1 -2 -3 -3 -3 0 -2 -3 -2 -2 -1 -1 -3 0 0 -1 -2 -3 -2 6 -2 -4 -4 -1 -2 -2 -1 -1 -3 -3 -3 -3 -2 -2 -3 2 -2 -1 -1 0 -2 -2 -2 0 -2 -1 -3 -2 -1 -2 -3 -1 -2 -1 -1 -3 -4 -2 1 3 -3 P -3 -1 -4 -3 -4 -1 -3 -3 -3 -3 -1 -1 -3 -1 -1 -1 S -2 0 -3 -2 -3 1 -3 -2 -3 -3 1 1 -3 1 3 1 T -1 -1 -3 0 -3 0 -1 -1 -1 -1 0 0 -2 -1 0 0 W -2 -3 12 -3 12 -3 -2 -2 -2 -2 -3 -3 7 -3 -3 -3 Y -1 -2 2 -1 2 -2 -1 0 -1 -1 -2 -2 0 -3 -2 -2 V 1 -3 -3 4 -3 0 1 3 2 1 0 0 0 -1 -2 -1 -1 4 1 -3 -2 0 -2 -3 -1 1 5 -3 -4 -3 -3 12 -3 -2 -2 2 -1 1 0 -3 -2 -3 -2 2 7 -2 -2 -4 0 -3 -1 0 1 M 2 K 3 W 4 V 5 W 6 A 7 L 8 L 9 L 10 L 11 A 12 A 13 W 14 A 15 A 16 A ... 37 S 38 G 39 T 40 W 41 Y 42 A A -1 -1 -3 0 -3 5 -2 -1 -1 -2 5 5 -2 3 2 4 R -2 1 -3 -3 -3 -2 -2 -3 -3 -2 -2 -2 -3 -2 -1 -2 N -2 0 -4 -3 -4 -2 -4 -3 -4 -4 -2 -2 -4 -1 0 -1 D -3 1 -5 -4 -5 -2 -4 -4 -4 -4 -2 -2 -4 -2 -1 -2 C -2 -4 -3 -1 -3 -1 -1 -1 -1 -1 -1 -1 -2 -1 -2 -1 Q -1 2 -2 -3 -2 -1 -2 -3 -2 -2 -1 -1 -2 -1 2 -1 E -2 4 -3 -3 -3 -1 -3 -3 -3 -3 -1 -1 -3 -2 0 -1 G -3 -2 -3 -4 -3 0 -4 -4 -4 -4 0 0 -4 4 2 3 H -2 0 -3 -4 -3 -2 -3 -3 -3 -3 -2 -2 -3 -2 -1 -2 I 1 -3 -3 3 -3 -2 2 2 2 2 -2 -2 1 -2 -3 -2 L 2 -3 -2 1 -2 -2 4 2 4 4 -2 -2 4 -2 -3 -2 K -2 3 -3 -3 -3 -1 -3 -3 -3 -3 -1 -1 -3 -1 0 -1 M 6 -2 -2 1 -2 -1 2 1 2 2 -1 -1 2 -2 -2 -1 F 0 -4 1 -1 1 -3 0 3 0 0 -3 -3 1 -3 -3 -3 P -3 -1 -4 -3 -4 -1 -3 -3 -3 -3 -1 -1 -3 -1 -1 -1 S -2 0 -3 -2 -3 1 -3 -2 -3 -3 1 1 -3 1 3 1 T -1 -1 -3 0 -3 0 -1 -1 -1 -1 0 0 -2 -1 0 0 W -2 -3 12 -3 12 -3 -2 -2 -2 -2 -3 -3 7 -3 -3 -3 Y -1 -2 2 -1 2 -2 -1 0 -1 -1 -2 -2 0 -3 -2 -2 V 1 -3 -3 4 -3 0 1 3 2 1 0 0 0 -1 -2 -1 2 0 0 -3 -2 4 -1 -3 -1 -3 -2 -2 0 -1 0 -4 -2 -2 -1 -2 -1 -5 -3 -2 -1 -3 -1 -3 -3 -1 0 -2 -1 -2 -2 -1 0 0 -1 -2 -3 -2 6 -2 -4 -4 -1 -2 -2 -1 -1 -3 -3 -3 -3 -2 -2 -3 2 -2 -1 -1 0 -2 -2 -2 0 -2 -1 -3 -2 -1 -2 -3 -1 -2 -1 -1 -3 -4 -2 1 3 -3 -1 4 1 -3 -2 0 -2 -3 -1 1 5 -3 -4 -3 -3 12 -3 -2 -2 2 -1 1 0 -3 -2 -3 -2 2 7 -2 -2 -4 0 -3 -1 0 1 M 2 K 3 W 4 V 5 W 6 A 7 L 8 L 9 L 10 L 11 A 12 A 13 W 14 A 15 A 16 A ... 37 S 38 G 39 T 40 W 41 Y 42 A A -1 -1 -3 0 -3 5 -2 -1 -1 -2 5 5 -2 3 2 4 R -2 1 -3 -3 -3 -2 -2 -3 -3 -2 -2 -2 -3 -2 -1 -2 N -2 0 -4 -3 -4 -2 -4 -3 -4 -4 -2 -2 -4 -1 0 -1 D -3 1 -5 -4 -5 -2 -4 -4 -4 -4 -2 -2 -4 -2 -1 -2 C Q E G H I L K M -2 -1 -2 -3 -2 1 2 -2 6 -4 2 4 -2 0 -3 -3 3 -2 -3 -2 -3 -3 -3 -3 -2 -3 -2 -1 -3 -3 -4 -4 3 1 -3 1 -3 -2 -3 -3 -3 -3 -2 -3 -2 -1 -1 -1 0 -2 -2 -2 -1 -1 -1 -2 -3 -4 -3 2 4 -3 2 -1 -3 -3 -4 -3 2 2 -3 1 -1 -2 that -3 -4a -3 2 amino 4 -3 2 note given -1 -2 -3 -4 -3 2 4 -3 2 acid -1 -1 (such -1 0 as -2alanine) -2 -2 -1in-1 -1 -1 query -1 0 -2 -2 -2 can -1 -1 your protein -2 -2 -3 -4 -3 1 4 -3 2 receive -1 -1 -2 different 4 -2 -2 scores -2 -1 -2 -2 0 2 -1alanine— -3 -3 0 -2 for 2matching -1 -1 -1 3 -2 -2 -2 -1 -1 F 0 -4 1 -1 1 -3 0 3 0 0 -3 -3 1 -3 -3 -3 P -3 -1 -4 -3 -4 -1 -3 -3 -3 -3 -1 -1 -3 -1 -1 -1 W -2 -3 12 -3 12 -3 -2 -2 -2 -2 -3 -3 7 -3 -3 -3 Y -1 -2 2 -1 2 -2 -1 0 -1 -1 -2 -2 0 -3 -2 -2 V 1 -3 -3 4 -3 0 1 3 2 1 0 0 0 -1 -2 -1 2 0 0 -3 -2 4 -1 -3 -1 -3 -2 -2 0 -1 0 -4 -2 -2 -1 -2 -1 -5 -3 -2 -2 -3 -1 -2 -1 -1 -3 -4 -2 1 3 -3 -1 4 1 -3 -2 0 -2 -3 -1 1 5 -3 -4 -3 -3 12 -3 -2 -2 2 -1 1 0 -3 -2 -3 -2 2 7 -2 -2 -4 0 -3 -1 0 depending on the -1 0 0 in 0 the -1 -2 -3 0 position protein -3 -1 -3 -3 -1 -2 -1 -2 -2 -1 -2 6 -2 -4 -4 -1 -2 -2 -1 -1 -3 -3 -3 -3 -2 -2 -3 2 -2 -1 -1 0 -2 -2 -2 -2 -1 -3 -2 -1 S -2 0 -3 -2 -3 1 -3 -2 -3 -3 1 1 -3 1 3 1 T -1 -1 -3 0 -3 0 -1 -1 -1 -1 0 0 -2 -1 0 0 1 M 2 K 3 W 4 V 5 W 6 A 7 L 8 L 9 L 10 L 11 A 12 A 13 W 14 A 15 A 16 A ... 37 S 38 G 39 T 40 W 41 Y 42 A A -1 -1 -3 0 -3 5 -2 -1 -1 -2 5 5 -2 3 2 4 R -2 1 -3 -3 -3 -2 -2 -3 -3 -2 -2 -2 -3 -2 -1 -2 N -2 0 -4 -3 -4 -2 -4 -3 -4 -4 -2 -2 -4 -1 0 -1 D -3 1 -5 -4 -5 -2 -4 -4 -4 -4 -2 -2 -4 -2 -1 -2 2 0 0 -3 -2 4 -1 -3 -1 -3 -2 -2 0 -1 0 -4 -2 -2 -1 -2 -1 -5 -3 -2 C Q E G H I L K M -2 -1 -2 -3 -2 1 2 -2 6 -4 2 4 -2 0 -3 -3 3 -2 -3 -2 -3 -3 -3 -3 -2 -3 -2 -1 -3 -3 -4 -4 3 1 -3 1 -3 -2 -3 -3 -3 -3 -2 -3 -2 -1 -1 -1 0 -2 -2 -2 -1 -1 -1 -2 -3 -4 -3 2 4 -3 2 -1 -3 -3 -4 -3 2 2 -3 1 note a given -1 -2 that -3 -4 -3 2 amino 4 -3 2 -1 -2(such -3 -4as-3tryptophan) 2 4 -3 2 acid -1 -1 -1 0 -2 -2 -2 -1 -1 in -1 your -1 -1query 0 -2 protein -2 -2 -1can -1 -2 -2 -3different -4 -3 1scores 4 -3 2 receive -1 -1 -2 4 -2 -2 -2 -1 -2 for -2 matching 2 0 2 -1 -3 -3 0 -2 -1 -1 -1 3 -2 -2 -2 -1 -1 tryptophan—depending on in -3 the 0 -1 the 0 0position 0 -1 -2 -3 -2 -2 6 -2 -4 -4 -2 protein -1 -3 -3 -1 -1 -2 -2 -1 -1 -2 -2 -1 -1 -3 -3 -3 -3 -2 -2 -3 2 -2 -1 -1 0 -2 -2 -2 -1 -3 -2 -1 -2 -3 -1 -2 -1 -1 F 0 -4 1 -1 1 -3 0 3 0 0 -3 -3 1 -3 -3 -3 P -3 -1 -4 -3 -4 -1 -3 -3 -3 -3 -1 -1 -3 -1 -1 -1 S -2 0 -3 -2 -3 1 -3 -2 -3 -3 1 1 -3 1 3 1 T -1 -1 -3 0 -3 0 -1 -1 -1 -1 0 0 -2 -1 0 0 W -2 -3 12 -3 12 -3 -2 -2 -2 -2 -3 -3 7 -3 -3 -3 Y -1 -2 2 -1 2 -2 -1 0 -1 -1 -2 -2 0 -3 -2 -2 V 1 -3 -3 4 -3 0 1 3 2 1 0 0 0 -1 -2 -1 -3 -4 -2 1 3 -3 -1 4 1 -3 -2 0 -2 -3 -1 1 5 -3 -4 -3 -3 12 -3 -2 -2 2 -1 1 0 -3 -2 -3 -2 2 7 -2 -2 -4 0 -3 -1 0 PSI-BLAST is performed in five steps [1] Select a query and search it against a protein database [2] PSI-BLAST constructs a multiple sequence alignment then creates a “profile” or specialized position-specific scoring matrix (PSSM) [3] The PSSM is used as a query against the database B&FG 3e Page 172 PSI-BLAST is performed in five steps [1] Select a query and search it against a protein database [2] PSI-BLAST constructs a multiple sequence alignment then creates a “profile” or specialized position-specific scoring matrix (PSSM) [3] The PSSM is used as a query against the database [4] PSI-BLAST estimates statistical significance (E values) B&FG 3e Page 172 PSI-BLAST is performed in five steps [1] Select a query and search it against a protein database [2] PSI-BLAST constructs a multiple sequence alignment then creates a “profile” or specialized position-specific scoring matrix (PSSM) [3] The PSSM is used as a query against the database [4] PSI-BLAST estimates statistical significance (E values) [5] Repeat steps [3] and [4] iteratively, typically 5 times. At each new search, a new profile is used as the query. B&FG 3e Page 172 Position-specific scoring matrix (PSSM) B&FG 3e Fig. 5.3 Page 173 PSI-BLAST: dramatic increase in number of hits B&FG 3e Table 5.2 Page 174 Given this query, a standard BLASTP search would produce about 9 hits with low expect values. This PSI-BLAST search produces >200 hits after 3 or 4 iterations. Note that PSI-BLAST E values can improve dramatically! After 1st iteration: Expect = 4e-04 Alignment length = 87 amino acids After 2nd iteration: Expect = 1e-36 Alignment length = 110 amino acids After 3rd iteration: Expect = 2e-33 Alignment length = 146 amino acids B&FG 3e Fig. 5.4 Page 175 The universe of lipocalins (each dot is a protein) retinol-binding protein apolipoprotein D odorant-binding protein Scoring matrices let you focus on the big (or small) picture retinol-binding protein your RBP query Scoring matrices let you focus on the big (or small) picture PAM250 PAM30 retinol-binding retinol-binding protein protein Blosum80 Blosum45 PSI-BLAST generates scoring matrices more sensitive than PAM or BLOSUM retinol-binding protein PSI‐BLAST algorithm increases the sensitivity of a database search by detecting homologous matches with relatively low sequence identity B&FG 3e Fig. 5.5 Page 176 PSI-BLAST: the problem of corruption In PSI-BLAST once a match is incorporated into a PSSM it will never be removed, even if it is wrong (i.e. even if it is a false positive that is not truly homologous to the query). Not only will it stay, it may lead to the inclusion of many other related false positive hits. There are three main approaches to removing false positives: B&FG 3e Page 177 (1) Filter biased amino acid regions. (This is an option in BLAST.) (2) Lower the expect value threshold to make the search more stringent. (3) Visually inspect the output from each PSI-BLAST iteration and remove suspicious matches (by unchecking the corresponding boxes).