
No Slide Title
... • To introduce the theory and practice of phylogenetic inference from molecular data ...
... • To introduce the theory and practice of phylogenetic inference from molecular data ...

Evolution/Phylogeny
... 1. Make star tree with ‘fake’ distances (we need these to be able to calculate total branch length) 2. Check all n(n-1)/2 possible pairs and join the pair that leads to smallest total branch length. You do this for each pair by calculating the real branch lengths from the pair to the common ancestor ...
... 1. Make star tree with ‘fake’ distances (we need these to be able to calculate total branch length) 2. Check all n(n-1)/2 possible pairs and join the pair that leads to smallest total branch length. You do this for each pair by calculating the real branch lengths from the pair to the common ancestor ...

From phylogenetic trees to networks
... evolutionary history of genes, populations, species. They are typically reconstructed with a wide range of algorithms from the comparison of very long strings representing the molecular (DNA or protein) sequences found across different organisms. Phylogenetic reconstruction is part of the daily rout ...
... evolutionary history of genes, populations, species. They are typically reconstructed with a wide range of algorithms from the comparison of very long strings representing the molecular (DNA or protein) sequences found across different organisms. Phylogenetic reconstruction is part of the daily rout ...

L1KProcs: an R package for L1000 data processing and analysis
... • L1KProcs is an R package and interface for LINCS L1000 data preprocessing and compound signature detection in both textmode and graphic-mode way. • Additionally, it is a library for existing L1000 processed expression data and their connections (EGEM library). ...
... • L1KProcs is an R package and interface for LINCS L1000 data preprocessing and compound signature detection in both textmode and graphic-mode way. • Additionally, it is a library for existing L1000 processed expression data and their connections (EGEM library). ...

Document
... • The 3/4 and 4/3 terms reflect that there are four types of nucleotides and three ways in which a second nucleotide may not match a first - with all types of change being equally likely (i.e. unrelated sequences should be 25% identical by chance alone) ...
... • The 3/4 and 4/3 terms reflect that there are four types of nucleotides and three ways in which a second nucleotide may not match a first - with all types of change being equally likely (i.e. unrelated sequences should be 25% identical by chance alone) ...

A Platform for Cluster Analysis of Next
... The purpose of gene expression data clustering analysis is clustered genes with the same or similar functions to help explore the gene function and regulatory network. The past is mainly based on microarray gene expression data, in recent years due to the development of next-generation sequencing te ...
... The purpose of gene expression data clustering analysis is clustered genes with the same or similar functions to help explore the gene function and regulatory network. The past is mainly based on microarray gene expression data, in recent years due to the development of next-generation sequencing te ...

A Bayesian Method for Rank Agreggation
... The method is usually applied to rank the top K candidates, so P is KK matrix ◦ Let U be the list of genes that hare ranked as top K at least once in some experiment ◦ For each pair of genes in U, let mi , j 1 if for a majority of experiments i is ranked above j. ◦ Define Pi , j mi , j / | U |, ...
... The method is usually applied to rank the top K candidates, so P is KK matrix ◦ Let U be the list of genes that hare ranked as top K at least once in some experiment ◦ For each pair of genes in U, let mi , j 1 if for a majority of experiments i is ranked above j. ◦ Define Pi , j mi , j / | U |, ...

S6. Phylogenetic results: complementary analyses Bayesian
... S6. Phylogenetic results: complementary analyses Bayesian Inference analyses with MrBayes 1.2, with characteristics as described above for the main analyses, were also carried out under different partition schemes to understand whether these would influence the general topology of the Madascincus ph ...
... S6. Phylogenetic results: complementary analyses Bayesian Inference analyses with MrBayes 1.2, with characteristics as described above for the main analyses, were also carried out under different partition schemes to understand whether these would influence the general topology of the Madascincus ph ...

Trees
... Amino-acid sites are partially ordered characters. An amino acid cannot change into all other amino acids in a singe step, as sometimes 2 or 3 steps are required. For example, a tyrosine may only change into a leucine through an intermediate state, i.e., phenylalanine or histidine. ...
... Amino-acid sites are partially ordered characters. An amino acid cannot change into all other amino acids in a singe step, as sometimes 2 or 3 steps are required. For example, a tyrosine may only change into a leucine through an intermediate state, i.e., phenylalanine or histidine. ...


File
... The main idea of decision tree construction tree is to evaluate different attributes and different partitioning conditions, and pick the attributes and partitioning condition that results in the maximum information gain ratio. The same procedure works recursively on each of the sets resulting from t ...
... The main idea of decision tree construction tree is to evaluate different attributes and different partitioning conditions, and pick the attributes and partitioning condition that results in the maximum information gain ratio. The same procedure works recursively on each of the sets resulting from t ...

Hierarchical Clustering in R
... • How to find out what packages are actually installed locally – (.packages()) ...
... • How to find out what packages are actually installed locally – (.packages()) ...

KS3 curriculum links (England)
... heredity as the process by which genetic information is transmitted from one generation to the next a simple model of chromosomes, genes and DNA in heredity, including the part played by Watson, Crick, Wilkins and Franklin in the development of the DNA model differences between species the variation ...
... heredity as the process by which genetic information is transmitted from one generation to the next a simple model of chromosomes, genes and DNA in heredity, including the part played by Watson, Crick, Wilkins and Franklin in the development of the DNA model differences between species the variation ...

Exploratory Data Analysis Tools for Phylogenetics: Visualizing
... and some misleading (e.g. long branch attraction, compositional bias, changing patterns of variable sites). ...
... and some misleading (e.g. long branch attraction, compositional bias, changing patterns of variable sites). ...

Slajd 1
... The construction of phylogenetic trees from numerical methods The principle of maximum parsimony (Occam’s razor) holds that we should accept that phylogenetic tree that can be constructed with the least number of morphological changes. The raw data Species A B C D E ...
... The construction of phylogenetic trees from numerical methods The principle of maximum parsimony (Occam’s razor) holds that we should accept that phylogenetic tree that can be constructed with the least number of morphological changes. The raw data Species A B C D E ...

tree - Tecfa
... Maximizes the likelihood of observing the sequence data for a specific model of character state changes Likelihood of a site = Sum of probabilities of every possible reconstruction of ancestral states at the internal nodes Likelyhood of the tree = Product of the likelihoods for all sites (=sum of lo ...
... Maximizes the likelihood of observing the sequence data for a specific model of character state changes Likelihood of a site = Sum of probabilities of every possible reconstruction of ancestral states at the internal nodes Likelyhood of the tree = Product of the likelihoods for all sites (=sum of lo ...
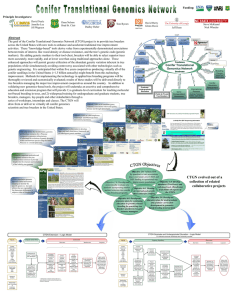
Objective 2.0
... markers). By adding genetic markers to their tool-chest, breeders will be able to select superior trees more accurately, more rapidly, and at lower cost than using traditional approaches alone. These enhanced approaches will permit greater utilization of the abundant genetic variation inherent in tr ...
... markers). By adding genetic markers to their tool-chest, breeders will be able to select superior trees more accurately, more rapidly, and at lower cost than using traditional approaches alone. These enhanced approaches will permit greater utilization of the abundant genetic variation inherent in tr ...

Global Similarity Between Multiple Bionetworks
... Sequence sorting: The nodes are generally weighted by different attributes; however, the occurrence of same nodes in graphs greatly increase the complexity for finding the maximal global similarities between two networks. Transitivity: Especially in functional networks, the transitivity should b ...
... Sequence sorting: The nodes are generally weighted by different attributes; however, the occurrence of same nodes in graphs greatly increase the complexity for finding the maximal global similarities between two networks. Transitivity: Especially in functional networks, the transitivity should b ...

Networks: expanding evolutionary thinking
... data carry the promise of fascinating insights into treelike processes, non-treelike processes are commonly observed. Network analysis is a readily available and ideal tool that reduces the danger of misinterpreting such data. Tackling error with networks There are long-standing controversies regard ...
... data carry the promise of fascinating insights into treelike processes, non-treelike processes are commonly observed. Network analysis is a readily available and ideal tool that reduces the danger of misinterpreting such data. Tackling error with networks There are long-standing controversies regard ...
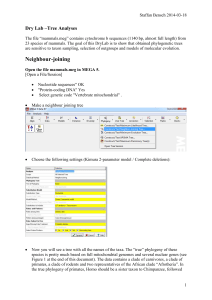
Dry Lab – More Tree Analayses
... Question 3. Which clades have high support? Are there any inconsistencies relative the “true tree” that have good support? ...
... Question 3. Which clades have high support? Are there any inconsistencies relative the “true tree” that have good support? ...

View the seminar poster
... totalling more than 700 species worldwide. Subjected to phylogene5c analysis since the late 1990s, early studies drew on small sets of external morphological characters, mostly those used in classical ...
... totalling more than 700 species worldwide. Subjected to phylogene5c analysis since the late 1990s, early studies drew on small sets of external morphological characters, mostly those used in classical ...

Bioinformatics and Supercomputing
... action of the Alu ‘restriction’ endonucleous. •Discovery of Alu subfamillies led to hypothesis of master/ source genes. AGCT •Reveal ancestry because individuals only share particular sequence insertion if the share an ancestor. •Can identify similarities of functional, structural, or evolutionary r ...
... action of the Alu ‘restriction’ endonucleous. •Discovery of Alu subfamillies led to hypothesis of master/ source genes. AGCT •Reveal ancestry because individuals only share particular sequence insertion if the share an ancestor. •Can identify similarities of functional, structural, or evolutionary r ...

Document
... ClustalW and Phylip do bootstrap operations automatically Bootstrapping involves these steps: ...
... ClustalW and Phylip do bootstrap operations automatically Bootstrapping involves these steps: ...


Intraspecific gene genealogies: trees grafting into networks
... network is built by taking these splits and combining them successively when splits are incompatible, a loop is introduced to indicate that there are alternative splits fast, nucleotide or protein data, allows for the inclusion of models of nucleotide substitution or amino acid replacement, bootstra ...
... network is built by taking these splits and combining them successively when splits are incompatible, a loop is introduced to indicate that there are alternative splits fast, nucleotide or protein data, allows for the inclusion of models of nucleotide substitution or amino acid replacement, bootstra ...