
Ming Li Talk about Bioinformatics - the David R. Cheriton School of
... Hence at least in the past they had the same structure and function. Caution: old genes can be recruited for new functions. Example: a structural protein in eye lens is homologous to an ancient glycolytic enzyme. ...
... Hence at least in the past they had the same structure and function. Caution: old genes can be recruited for new functions. Example: a structural protein in eye lens is homologous to an ancient glycolytic enzyme. ...

How are the proteins built up
... proteins) fold into compact structures, and the length of the secondary “modules” are limited and connected by “loops” in order to form these globular structures. In general, the way proteins fold is thought to proceed in steps and along certain paths so that the catastrophe of the Leventhal paradox ...
... proteins) fold into compact structures, and the length of the secondary “modules” are limited and connected by “loops” in order to form these globular structures. In general, the way proteins fold is thought to proceed in steps and along certain paths so that the catastrophe of the Leventhal paradox ...

Quaternary structures
... Changes in shape are an important part of protein function and control. For example: a change in shape allows DNA methyltransferase to choose hemi-methylated meCG/GC for bimethylation to meCG/GmeC ...
... Changes in shape are an important part of protein function and control. For example: a change in shape allows DNA methyltransferase to choose hemi-methylated meCG/GC for bimethylation to meCG/GmeC ...

PPT (without movies)
... important scientific research. Join this free online game and help us predict the folds of unsolved proteins as well as designing new proteins to cure diseases. We’re collecting data to find out if humans' pattern-recognition and puzzle-solving abilities make them more efficient than existing comput ...
... important scientific research. Join this free online game and help us predict the folds of unsolved proteins as well as designing new proteins to cure diseases. We’re collecting data to find out if humans' pattern-recognition and puzzle-solving abilities make them more efficient than existing comput ...

Usha`s project - The University of Texas at Dallas
... DALI (Distance Matrix Alignment; Holm and Sander 1993a) uses distance matrices to represent each protein structure. In this method, each structure is represented as a two-dimensional array of distances between all the C-alpha atoms. Figure 19 illustrates how a distance matrix is obtained for a prote ...
... DALI (Distance Matrix Alignment; Holm and Sander 1993a) uses distance matrices to represent each protein structure. In this method, each structure is represented as a two-dimensional array of distances between all the C-alpha atoms. Figure 19 illustrates how a distance matrix is obtained for a prote ...

ProteinChipâ technology is one of the most exciting advancements
... ProteinChip technology is one of the most exciting advancements in protein analysis in the last 5 years. The Protein Biology SystemTM (PBS) combines the power of mass analysis with chromatography surfaces on an integrated platform. The PBS can easily be used by biologists, biochemists, and clinicia ...
... ProteinChip technology is one of the most exciting advancements in protein analysis in the last 5 years. The Protein Biology SystemTM (PBS) combines the power of mass analysis with chromatography surfaces on an integrated platform. The PBS can easily be used by biologists, biochemists, and clinicia ...

Proteins - Boardworks
... The R group represents a side chain from the central “alpha” carbon atom, and can be anything from a simple hydrogen atom to a more complex ring structure. 3 of 8 ...
... The R group represents a side chain from the central “alpha” carbon atom, and can be anything from a simple hydrogen atom to a more complex ring structure. 3 of 8 ...

A central problem in bioinformatics
... Nucleotide sequence databanks contain 16 x 109 bases The full three-dimensional coordinates of proteins of average length ~400 residues: 16000 entries Not only are the individual databanks large, but their sizes are increasing as a very high rate. ...
... Nucleotide sequence databanks contain 16 x 109 bases The full three-dimensional coordinates of proteins of average length ~400 residues: 16000 entries Not only are the individual databanks large, but their sizes are increasing as a very high rate. ...

Structure Determination and Sequence Analysis - Rose
... Proteins exhibiting a high degree of sequence similarity (or greater than ~10 to 15% sequence identity) are usually considered to be evolutionarily-related. Evolutionarily related proteins exhibit similar structures and similar functions. Thus, by comparison of sequences of newly discovered proteins ...
... Proteins exhibiting a high degree of sequence similarity (or greater than ~10 to 15% sequence identity) are usually considered to be evolutionarily-related. Evolutionarily related proteins exhibit similar structures and similar functions. Thus, by comparison of sequences of newly discovered proteins ...


Teaching Notes
... localization. In 2008 Shimomura, Chalfie and Tsien were awarded Nobel Prize in Chemistry for their contributions to this field. Green Fluorescent Protein has been widely used as a tracking reagent in biology and biotechnology. The protein itself is shaped like a barrel made of a single beta sheet. I ...
... localization. In 2008 Shimomura, Chalfie and Tsien were awarded Nobel Prize in Chemistry for their contributions to this field. Green Fluorescent Protein has been widely used as a tracking reagent in biology and biotechnology. The protein itself is shaped like a barrel made of a single beta sheet. I ...

structbio_lecture_BCH391L_20150212.ppt
... Comparative Modeling – create a structure model for a protein of interest ...
... Comparative Modeling – create a structure model for a protein of interest ...

6th semester-2006 Project Proposal
... “Construction of plasmid vectors to tag proteins for universal light-induced protein immobilization on surfaces” Background: A method of light-induced immobilization of proteins(1,2) on chemically treated surfaces has been successfully developed over the past years in the group, by Teresa Petersen a ...
... “Construction of plasmid vectors to tag proteins for universal light-induced protein immobilization on surfaces” Background: A method of light-induced immobilization of proteins(1,2) on chemically treated surfaces has been successfully developed over the past years in the group, by Teresa Petersen a ...
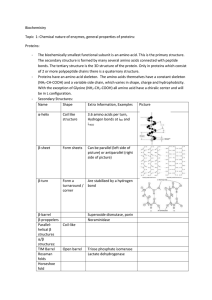
Biochemistry Topic 1: Chemical nature of enzymes, general
... Membrane Proteins have a sequence of hydrophobic amino acids that will be inside the lipid bilayer membrane as the phospholipids are hydrophobic. Some will cross the membrane several times others only once. Cytosolic proteins are membrane proteins which do not cross the membrane but are only attache ...
... Membrane Proteins have a sequence of hydrophobic amino acids that will be inside the lipid bilayer membrane as the phospholipids are hydrophobic. Some will cross the membrane several times others only once. Cytosolic proteins are membrane proteins which do not cross the membrane but are only attache ...

Nitrogen Balance
... • If the diet is deficient in lipid, then a greater proportion of dietary protein is metabolized for energy or deaminated for conversion to fat and carbohydrate, more ammonia is excreted, and a lower percentage of dietary N is retained for growt h ...
... • If the diet is deficient in lipid, then a greater proportion of dietary protein is metabolized for energy or deaminated for conversion to fat and carbohydrate, more ammonia is excreted, and a lower percentage of dietary N is retained for growt h ...

Structural Biology in the Pharmaceutical Industry
... Historically, the main contribution of protein crystallography was in the generation of crystal structures of the target protein in complex with lead molecules during the lead optimization process. This helped to speed up lead optimization by providing the medicinal chemists with the view of the 3D ...
... Historically, the main contribution of protein crystallography was in the generation of crystal structures of the target protein in complex with lead molecules during the lead optimization process. This helped to speed up lead optimization by providing the medicinal chemists with the view of the 3D ...

pptx
... The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation • Interactions between adjacent R-groups – Ionic attraction or repulsion – Steric hindrance of adjacent bulky groups ...
... The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation • Interactions between adjacent R-groups – Ionic attraction or repulsion – Steric hindrance of adjacent bulky groups ...

Understanding the complexity of Protein Function
... Background Information • Discovered over 20 years ago • Enzyme Class: Isomerase • Is a member of the cytochrome P450 enzyme family • P450s belong to a large family of proteins containing a heme cofactor • P450 enzymes have been identified in all domains of life • Prostaglandins are derivative of Ar ...
... Background Information • Discovered over 20 years ago • Enzyme Class: Isomerase • Is a member of the cytochrome P450 enzyme family • P450s belong to a large family of proteins containing a heme cofactor • P450 enzymes have been identified in all domains of life • Prostaglandins are derivative of Ar ...

DOC
... vivo. However, acting upstream the proteic level is a limiting factor if the turnover of the target protein is slow or the existing pool of the target protein is important (for instance, in insect embryos, as a consequence of a strong maternal contribution). In order to circumvent these problems, we ...
... vivo. However, acting upstream the proteic level is a limiting factor if the turnover of the target protein is slow or the existing pool of the target protein is important (for instance, in insect embryos, as a consequence of a strong maternal contribution). In order to circumvent these problems, we ...

Predicting protein 3D structure from evolutionary sequence variation
... these inferred couplings is an excellent predictor of residue-residue proximity in folded structures. Indeed, the top-scoring residue couplings are sufficiently accurate and well-distributed to define the 3D protein fold with remarkable accuracy. We quantify this observation by computing, from seque ...
... these inferred couplings is an excellent predictor of residue-residue proximity in folded structures. Indeed, the top-scoring residue couplings are sufficiently accurate and well-distributed to define the 3D protein fold with remarkable accuracy. We quantify this observation by computing, from seque ...

Basic Local Alignment Search Tool
... Motif Prediction Restriction Analysis (TCGA) RNAFOLD (GCG) ...
... Motif Prediction Restriction Analysis (TCGA) RNAFOLD (GCG) ...


Slides
... Protein : adrendoxin (Adx) • In mitochondria of the adrenal cortex, the steroid hydroxylating system requires the transfer of electrons from the membrane-attached flavoprotein AR via the soluble Adx to the membrane-integrated cytochrome P450 of the CYP 11 family. ...
... Protein : adrendoxin (Adx) • In mitochondria of the adrenal cortex, the steroid hydroxylating system requires the transfer of electrons from the membrane-attached flavoprotein AR via the soluble Adx to the membrane-integrated cytochrome P450 of the CYP 11 family. ...

No Slide Title
... • PROSITE provides consensus patterns for a lot of PTM sites, however in most cases these patterns are very short and the true modifications occur based on the structural or environmental context in the protein fold • Because of this reason, methods based on reg expressions or local alignment method ...
... • PROSITE provides consensus patterns for a lot of PTM sites, however in most cases these patterns are very short and the true modifications occur based on the structural or environmental context in the protein fold • Because of this reason, methods based on reg expressions or local alignment method ...
Structural alignment

Structural alignment attempts to establish homology between two or more polymer structures based on their shape and three-dimensional conformation. This process is usually applied to protein tertiary structures but can also be used for large RNA molecules. In contrast to simple structural superposition, where at least some equivalent residues of the two structures are known, structural alignment requires no a priori knowledge of equivalent positions. Structural alignment is a valuable tool for the comparison of proteins with low sequence similarity, where evolutionary relationships between proteins cannot be easily detected by standard sequence alignment techniques. Structural alignment can therefore be used to imply evolutionary relationships between proteins that share very little common sequence. However, caution should be used in using the results as evidence for shared evolutionary ancestry because of the possible confounding effects of convergent evolution by which multiple unrelated amino acid sequences converge on a common tertiary structure.Structural alignments can compare two sequences or multiple sequences. Because these alignments rely on information about all the query sequences' three-dimensional conformations, the method can only be used on sequences where these structures are known. These are usually found by X-ray crystallography or NMR spectroscopy. It is possible to perform a structural alignment on structures produced by structure prediction methods. Indeed, evaluating such predictions often requires a structural alignment between the model and the true known structure to assess the model's quality. Structural alignments are especially useful in analyzing data from structural genomics and proteomics efforts, and they can be used as comparison points to evaluate alignments produced by purely sequence-based bioinformatics methods.The outputs of a structural alignment are a superposition of the atomic coordinate sets and a minimal root mean square deviation (RMSD) between the structures. The RMSD of two aligned structures indicates their divergence from one another. Structural alignment can be complicated by the existence of multiple protein domains within one or more of the input structures, because changes in relative orientation of the domains between two structures to be aligned can artificially inflate the RMSD.