
Pupko_pairwise
... based on the catalytic domain of: (a) 70 known ePKs from yeast, worm, fly, and human with > 50% identity in the ePK domain (b) each subfamily of known aPKs • HMM-profile searches and PSI-BLAST searches were performed ...
... based on the catalytic domain of: (a) 70 known ePKs from yeast, worm, fly, and human with > 50% identity in the ePK domain (b) each subfamily of known aPKs • HMM-profile searches and PSI-BLAST searches were performed ...

PPT
... alignment using a modified Needleman–Wunsch algorithm. After the sequence or secondary structure alignment is complete, SuperPose then generates a difference distance (DD) matrix between aligned alpha carbon atoms. A difference distance matrix can be generated by first calculating the distances betw ...
... alignment using a modified Needleman–Wunsch algorithm. After the sequence or secondary structure alignment is complete, SuperPose then generates a difference distance (DD) matrix between aligned alpha carbon atoms. A difference distance matrix can be generated by first calculating the distances betw ...

Let`s Get Pumped Up about Proteins!!!
... • Biochemical compounds that contain nitrogen as well as C, H, O, and sometimes S. (Remember: CHNOS) • consist of 1 or more polypeptide chains folded and coiled into specific conformations • What is the difference between a polypeptide and a protein? ...
... • Biochemical compounds that contain nitrogen as well as C, H, O, and sometimes S. (Remember: CHNOS) • consist of 1 or more polypeptide chains folded and coiled into specific conformations • What is the difference between a polypeptide and a protein? ...

The molecular architecture, macro-organization and functions of the
... Light-harvesting complex II (LHCII), the major antenna pigment-protein complex of plants and green algae, is the most abundant membrane protein on Earth. The primary function of LHCII is to capture sunlight and transfer the excitation energy to the photochemical reaction centers – with up to nearly ...
... Light-harvesting complex II (LHCII), the major antenna pigment-protein complex of plants and green algae, is the most abundant membrane protein on Earth. The primary function of LHCII is to capture sunlight and transfer the excitation energy to the photochemical reaction centers – with up to nearly ...

Protein Structure - Chemistry Courses: About: Department
... 1. Which statement is false about a globular protein that performs its biological function as a single independent polypeptide chain? A) Its tertiary structure is likely stabilized by the interactions of amino acid side chains in non-neighboring regions of the polypeptide chain. B) It could contain ...
... 1. Which statement is false about a globular protein that performs its biological function as a single independent polypeptide chain? A) Its tertiary structure is likely stabilized by the interactions of amino acid side chains in non-neighboring regions of the polypeptide chain. B) It could contain ...

Sequence identity and homology
... a local alignment algorithm decide where to stop? By lengthening the alignment only insofar as it increases the score. For example, one could increase the score by +2 for every identical amino acid, while assigning a penalty of -1 for every mismatch or gap. Such penalties would prevent the alignment ...
... a local alignment algorithm decide where to stop? By lengthening the alignment only insofar as it increases the score. For example, one could increase the score by +2 for every identical amino acid, while assigning a penalty of -1 for every mismatch or gap. Such penalties would prevent the alignment ...

STRUCTURAL ORGANIZATION OF LIVING SYSTEMS At all levels
... an unfolded polypeptide chain, the molecule has an astronomical number of possible conformations. Let us consider a protein that is built of 100 amino acid residues. If we take, that each amino acid has 2 possible conformations, the whole polypeptide chain must have 2100 possible conformations. If t ...
... an unfolded polypeptide chain, the molecule has an astronomical number of possible conformations. Let us consider a protein that is built of 100 amino acid residues. If we take, that each amino acid has 2 possible conformations, the whole polypeptide chain must have 2100 possible conformations. If t ...

DN: Protein
... sequence of the 20 different amino acids as illustrated on the left. In the feed lab, protein is distinguishable from carbohydrate and lipid due to its content of nitrogen (N) feed proteins typically contain about 16% N. This property makes it possible to estimate the protein content of a feedstuff ...
... sequence of the 20 different amino acids as illustrated on the left. In the feed lab, protein is distinguishable from carbohydrate and lipid due to its content of nitrogen (N) feed proteins typically contain about 16% N. This property makes it possible to estimate the protein content of a feedstuff ...

Appendix 3 Assessment of the effects of the observed variants We
... Non-synonymous variants were assessed using the SNP prediction tools SIFT, PolyPhen2, SNPs3D, PMut and SNPs&GO. The algorithm SIFT (6) is based on the principles of protein evolution, and uses sequence homology to predict the degree of amino acid conservation in protein sequences which correlates w ...
... Non-synonymous variants were assessed using the SNP prediction tools SIFT, PolyPhen2, SNPs3D, PMut and SNPs&GO. The algorithm SIFT (6) is based on the principles of protein evolution, and uses sequence homology to predict the degree of amino acid conservation in protein sequences which correlates w ...

Bioinformatics-Theory
... algorithms; Pair wise sequence alignment - NEEDLEMAN and Wunsch, Smith Waterman algorithms; Multiple sequence alignments - CLUSTAL, PRAS; Patterns, motifs and Profiles in sequences. UNIT IV Protein Structure Prediction and Protein Folding: Protein architecture, classification of protein structures – ...
... algorithms; Pair wise sequence alignment - NEEDLEMAN and Wunsch, Smith Waterman algorithms; Multiple sequence alignments - CLUSTAL, PRAS; Patterns, motifs and Profiles in sequences. UNIT IV Protein Structure Prediction and Protein Folding: Protein architecture, classification of protein structures – ...

slides
... contacts in proteins leave an evolutionary record Although evolutionary couplings show promise for the identification of functional sites, homomultimer contacts, alternative conformations and functional sites, many of the predicted contacts involved in these protein features may appear as false posi ...
... contacts in proteins leave an evolutionary record Although evolutionary couplings show promise for the identification of functional sites, homomultimer contacts, alternative conformations and functional sites, many of the predicted contacts involved in these protein features may appear as false posi ...

BINF 630 – Lecture 4 Introduction to Probability
... significance of the search results alignments • FASTA uses alignment scores between unrelated sequences to calculate the parameters of the extreme value distribution • BLAST calculates estimates of the statistical parameters based on the socring matrix and sequence composition. ...
... significance of the search results alignments • FASTA uses alignment scores between unrelated sequences to calculate the parameters of the extreme value distribution • BLAST calculates estimates of the statistical parameters based on the socring matrix and sequence composition. ...

COS 597c: Topics in Computational Molecular Biology Lecturer: Mona Singh
... Broadly speaking, a sequence motif is a conserved element of a sequence alignment. Its function or structure may be known, or its significance may be unknown. Thus, one way to get functional or structural information about a sequence is to determine what motifs it contains. In a sense, we have alrea ...
... Broadly speaking, a sequence motif is a conserved element of a sequence alignment. Its function or structure may be known, or its significance may be unknown. Thus, one way to get functional or structural information about a sequence is to determine what motifs it contains. In a sense, we have alrea ...

PowerPoint - IBIVU - Vrije Universiteit Amsterdam
... What is the function of the new gene? The “lazy” investigation (i.e., no biologial experiments, just bioinformatics techniques): – Find a set of similar protein sequences to the unknown sequence ...
... What is the function of the new gene? The “lazy” investigation (i.e., no biologial experiments, just bioinformatics techniques): – Find a set of similar protein sequences to the unknown sequence ...

Episode 23 0 Proetin: Structure and Function
... 4. The video estimates that 100 billion proteins may exist. How can so many proteins form from just a few molecules? When amino acids are joined in different orders, different proteins result. 5. What is the name of the bond that results in the formation of proteins? Peptide bond 6. What are the nam ...
... 4. The video estimates that 100 billion proteins may exist. How can so many proteins form from just a few molecules? When amino acids are joined in different orders, different proteins result. 5. What is the name of the bond that results in the formation of proteins? Peptide bond 6. What are the nam ...

Secondary Structure Prediction Protein Folding
... A protein (which is a linear sequence of amino acids) folds into a unique 3D structure: determine this structure ...
... A protein (which is a linear sequence of amino acids) folds into a unique 3D structure: determine this structure ...

tutorial on carbohydrates
... 12. The polymer chains of glycosaminoglycans are widely spread apart and bind large amount of water. a. What 2 functional groups of the polymer make this binding of water possible? b. What type of binding is involved? 13. In glycoproteins, what are the 3 amino acids to which the carbohydrate groups ...
... 12. The polymer chains of glycosaminoglycans are widely spread apart and bind large amount of water. a. What 2 functional groups of the polymer make this binding of water possible? b. What type of binding is involved? 13. In glycoproteins, what are the 3 amino acids to which the carbohydrate groups ...

Protein Function and Classification
... Construction of protein signatures • Construction of a multiple sequence alignment (MSA) from characterised protein sequences. • Modelling the pattern of conserved amino acids at specific positions within a MSA. • Use these models to infer relationships with the characterised sequences ...
... Construction of protein signatures • Construction of a multiple sequence alignment (MSA) from characterised protein sequences. • Modelling the pattern of conserved amino acids at specific positions within a MSA. • Use these models to infer relationships with the characterised sequences ...


Module 5
... against databases of motifs and profiles, or indeed both. Some commonly used programmes are listed below: Pfam is a collection of multiple alignments and profile hidden Markov models of protein domain families, which is based on proteins from both SWISS-PROT and SP-TrEMBL. SMART (a Simple Modular Ar ...
... against databases of motifs and profiles, or indeed both. Some commonly used programmes are listed below: Pfam is a collection of multiple alignments and profile hidden Markov models of protein domain families, which is based on proteins from both SWISS-PROT and SP-TrEMBL. SMART (a Simple Modular Ar ...


Bioinformatic analysis of diverse protein superfamilies to
... etc. Large structure-based sequence alignments have been created for each superfamily. Remote evolutionary relatives were superimposed by structural comparison, while sequencebased alignments were assumed meaningful for closer homologs [8]. Systematic bioinformatic analysis of genomic and structural ...
... etc. Large structure-based sequence alignments have been created for each superfamily. Remote evolutionary relatives were superimposed by structural comparison, while sequencebased alignments were assumed meaningful for closer homologs [8]. Systematic bioinformatic analysis of genomic and structural ...

Structural Bioinformatics In this presentation……
... • It can be very difficult to find correct, biologicallymeaningful alignments of very distantly related protein sequences because they contain only a very small proportion of identical monomers • In such cases, structural information can help because evolution tends to change structure less • Superi ...
... • It can be very difficult to find correct, biologicallymeaningful alignments of very distantly related protein sequences because they contain only a very small proportion of identical monomers • In such cases, structural information can help because evolution tends to change structure less • Superi ...

Seminar_3 - Great Lakes Genomics Center
... Less Commonly found: Hydrophobic interfaces Interchain Disulfides ...
... Less Commonly found: Hydrophobic interfaces Interchain Disulfides ...
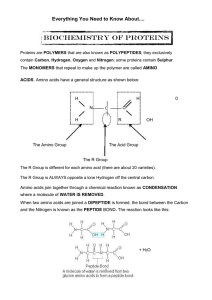
AS Biology - Everything Protein
... known as HYDROLYSIS. Protein molecules have very complex and intricate structures that let them perform their specific roles. PRIMARY STRUCTURE is the AMINO ACID SEQUENCE; peptide bonds are present in this level of structure. SECONDARY STRUCTURE is how the primary structure folds for the first time. ...
... known as HYDROLYSIS. Protein molecules have very complex and intricate structures that let them perform their specific roles. PRIMARY STRUCTURE is the AMINO ACID SEQUENCE; peptide bonds are present in this level of structure. SECONDARY STRUCTURE is how the primary structure folds for the first time. ...
Structural alignment

Structural alignment attempts to establish homology between two or more polymer structures based on their shape and three-dimensional conformation. This process is usually applied to protein tertiary structures but can also be used for large RNA molecules. In contrast to simple structural superposition, where at least some equivalent residues of the two structures are known, structural alignment requires no a priori knowledge of equivalent positions. Structural alignment is a valuable tool for the comparison of proteins with low sequence similarity, where evolutionary relationships between proteins cannot be easily detected by standard sequence alignment techniques. Structural alignment can therefore be used to imply evolutionary relationships between proteins that share very little common sequence. However, caution should be used in using the results as evidence for shared evolutionary ancestry because of the possible confounding effects of convergent evolution by which multiple unrelated amino acid sequences converge on a common tertiary structure.Structural alignments can compare two sequences or multiple sequences. Because these alignments rely on information about all the query sequences' three-dimensional conformations, the method can only be used on sequences where these structures are known. These are usually found by X-ray crystallography or NMR spectroscopy. It is possible to perform a structural alignment on structures produced by structure prediction methods. Indeed, evaluating such predictions often requires a structural alignment between the model and the true known structure to assess the model's quality. Structural alignments are especially useful in analyzing data from structural genomics and proteomics efforts, and they can be used as comparison points to evaluate alignments produced by purely sequence-based bioinformatics methods.The outputs of a structural alignment are a superposition of the atomic coordinate sets and a minimal root mean square deviation (RMSD) between the structures. The RMSD of two aligned structures indicates their divergence from one another. Structural alignment can be complicated by the existence of multiple protein domains within one or more of the input structures, because changes in relative orientation of the domains between two structures to be aligned can artificially inflate the RMSD.