Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Signal transduction wikipedia , lookup
Paracrine signalling wikipedia , lookup
Ribosomally synthesized and post-translationally modified peptides wikipedia , lookup
Gene expression wikipedia , lookup
Ancestral sequence reconstruction wikipedia , lookup
Expression vector wikipedia , lookup
Amino acid synthesis wikipedia , lookup
Biosynthesis wikipedia , lookup
G protein–coupled receptor wikipedia , lookup
Point mutation wikipedia , lookup
Magnesium transporter wikipedia , lookup
Genetic code wikipedia , lookup
Bimolecular fluorescence complementation wikipedia , lookup
Homology modeling wikipedia , lookup
Protein purification wikipedia , lookup
Metalloprotein wikipedia , lookup
Interactome wikipedia , lookup
Western blot wikipedia , lookup
Biochemistry wikipedia , lookup
Two-hybrid screening wikipedia , lookup
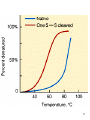
Major Problem in Modern Biochemistry “The Folding Problem” Background Information about the three dimensional structure of a protein is carried in the amino acid sequence- i.e. the gene. (Important concept) Early experiments Anfinsen - thermally denature ribonuclease (no cleavage) -refold the protein to be a functional enzyme How is the information encoded ? 1 Ribonuclease :Reversible folding & unfolding S-S bonds in brown Denaturation-studied by various methods is reversible 2 Problem:-The Lavinthal paradox Imagine a polypeptide chain of ~100 residues. Assume each amino acid can exist in 10 conformations. Therefore 10100 conformational states are possible. Each has a different set of thermodynamic properties How can the protein sample each state ? The number of states is 10 21 times greater than one estimate of the number of atoms in the universe. A protein hasn’t time to sample each conformation. Thus, folding must not be a completely random phenomenon. So-how can proteins fold ? 3 4 Second problem: The existence of a single conformation of a system that can exist in 10100 states is unlikely. Why ? Because in selecting a single state the conformational entropy S (conformational) = RlnN is lost. The unfavorable contribution to G is +RTlnN = 8.314 x 300 x ln 10100 = +574 kJ/mole So there must be stabilizing influences on H to bring the overall G to a minimum Thermodynamic parameters for the folding of some globular proteins at 25 C in aqueous solution First Issue: What features of protein folding produce large , negative H or large positive S changes, to compensate for the conformational entropy loss ? 5 Internal Interactions - energetically favorable interactions between groups within the folded molecule. 1. Charge-Charge Interactions - occur between positively and negatively charged side chain groups. 2. Internal Hydrogen Bonds - interactions between amino acid side chains that are either good hydrogen bond donors (such as the hydroxyls of serine or threonine) or good acceptors (such as the carbonyl oxygens of asparagine or glutamine) . Though hydrogen bonds are relatively weak, the large number of them can make a considerable contribution to stability. 3. van der Waals Interactions - weak interactions between uncharged molecular groups in the tightly packed environment of a folded protein. The contributions of these interactions to the negative enthalpy of folding is diminished by giving up favorable interactions with water via folding . 6 Common Errors - One of the most common folding errors occurs via cis-trans isomerization of the amide bond adjacent to a proline residue (see here). Proline is the only amino acid in proteins that forms peptide bonds in which the trans isomer is only slightly favored (4 to 1 versus 1000 to 1 for other residues). Thus, during folding, there is a significant chance that the wrong proline isomer will form first. It appears that cells have enzymes to catalyze the cis-trans isomerization necessary to speed correct folding. 7 DSC of lysozyme at 3 pH values. Differential Scanning calorimetry (DSC) is used to measure the heat required to raise the temperature of a sample at a constant rate, compared to a reference buffer. If the sample is undergoing a physical phase transition, some heat goes into causing the structural change so that more heat is needed to raise the temperature. T2 Δ H = Δ C p dt T1 where ΔC p is the difference in heat capacity between the sample and the buffer . 8 9 Dynamics of Protein Folding How do chains fold into their native structures in spite of astronomically large numbers of alternatives ? Clue: Existence of partially unfolded states High concentrations of Urea (8M) or guanidine-HCl (6M) leads to completely unfolded states of proteins. But- Other denaturing conditions lead to small changes in hydrodynamic and optical parameters. 1967:-John Brandts showed that acid and thermally denatures proteins can undergo another transition in guanidine-HCl Example-Bovine or human -lactalbumins undergo two different conformational transitions when guanidine is added. Characterization of different states of protein folding depends on the availability of a spectroscopic method. 10 NMR Spectroscopy simplified Nuclei of some elements have spin and behave like small magnets. The spin states are quantized (take on discrete values) Protons = 1/2, deuterons = 1, 13C = 1/2 (I is the number usually used for nuclear spin) There are 2I + 1 states for each nucleus. In an applied magnetic field, these substates have different energies and can be distinguished. 1H substates can have mI = -1/2 or +1/2 2H substates can have m = -1, 0 or +1 I Energy The energy of these substates in an applied magnetic (H) field is shown below. I= -1/2 E = difference in spin state energies I = +1/2 Magnetic field The spin of a nucleus can be flipped according to the equation: E = h. This occurs in the microwave region of the spectrum. Experimentally, either the microwave energy can be fixed and the magnetic field changed or vice versa. Normally the magnetic (H) field is swept. 11 The main use of NMR is derived from the fact that the energy levels of a spinning nucleus in a magnetic field depend on the atomic (electronic) environment. Different protons for example, absorb at different frequencies with respect to a reference material: = (Href -H)/ Href x 106 (ppm units) H = nucleus of interest Href = reference nucleus Spectacular advances in NMR result from the fact that nuclear spins also interact through space. Protons closer than 5 perturb each other’s spins (This is called the Nuclear Overhauser effect). This interaction allows determination of 3D structure in solution. > 6000 structures in the data base. Usually a “family” of structures is the solution for any single protein 12 13 14 Urea-induced unfolding of bovine carbonic anhydrase B (pH 3). At this pH the protein is in a molten globule state and unfolds by a one-step process. The fraction of unfolding is given by fu =(x-xo)/(xu- xo) where x is the value of the measured parameter, xo is its value in the absence of urea and xu is its value at high levels of urea. Symbols: , Relative intensity of trp fluorescence at 320/360 nm , increase in 1/P (P = trp fluorescence polarization) , decrease in the negative ellipticity at 220 nm increase in intrinsic viscosity Intrinsic viscosity and the spectrum and polarization of trp fluorescence reflect the compactness of the molecule, while the 220 nm ellipticity reflects secondary structure. 15 Unfolding of bovine carbonic anhydrase B at pH= 7.5 The fraction of unfolding is given by fu =(x-xo)/(xu- xo) where x is the value of the measured parameter, xo is its value in the absence of urea and xu is its value at high levels of urea. Symbols: Relative intensity increase in 1/P (P = trp fluorescence polarization) Increase in I(320)/I(360) x, , increase in signal intensities of aliphatic protons at 3.17, 2.97, 2.00, 0.86 and 1.38 ppm decrease in negative ellipticity at 270 nm Decrease in area under high field NMR resonance Fluorescence parameters reflect compactness of the molecule ellipticity at 220 reflects secondary structure area under high field resonance reflects tertiary structure 16 Spectroscopy shows the following: The first transition (pH 7.5) shows destruction of the rigid environment of the aromatic groups, The second transition shows destruction of the secondary structure The absence of rigid tertiary structure and the presence of secondary structure lead to a model of the intermediate state as possessing unfolded, non-compact molecules with local secondary structure. The state is termed the “molten globule” Properties of the molten globule I-Compactness . For -lactalbumin, the hydrodynamic radius is ~15% greater than the native state and the volume is 50% greater than the native state. The fully unfolded molecule (with S-S bonds) has a hydrodynamic radius increased by 49% from the native state and a volume increased by a factor of 3.3 from the native state. II. Presence of core. Non-polar groups are in contact but not as tightly as in the native protein III. Secondary structure. Similar to native 17 NMR can be used to determine the mobility of protein structure HD Exchange can be monitored in D2O solution Some internal H-bonded atoms can exchange quickly. Why ? Local unfolding or “breathing”. Either a normally buried group must surface occasionally to appear at the surface or the reagent must permeate to the interior. k1 Folded k2 open exchange k-1 k1 <<< k-1 18 Five hundred MHz proton NMR spectra of guinea pig -lactalbumin in the native (pH 5.4), acid (pH 2.0) and unfolded (in 9M urea) states recorded at 52oC. The NMR spectrum of the molten globule state is much simpler than that of the native protein;the number of perturbed resonances is much smaller, and the overall picture is much more similar to the unfolded state. 19 Time-dependence of the spin echo amplitudes for the methyl (A) and aromatic (B) protons of carbonic anhydrase B in the native (pH= 7.2) , molten globule (pH = 3.3) and unfolded (8M Urea) states. The spin-spin relaxation of the methyl groups in the molten globule state coincides with the unfolded state, while it is quite different for the native state. In contrast, the spin echo curves for the aromatic groups are intermediate between those in the native and in the unfolded states. Therefore, intramolecular movements of aromatic side chains are much more hindered in the molten globule than in the unfolded state, although not as hindered as in the native state. 20 IV: Native-like structural Organization NMR studies show that at least some -helices are located in their native positions along the polypeptide chain. How is this done: 1. Allow protein in the molten globule state to exchange for a given length of time. 2. Transform to native state 3. Use 2D-NMR to identify N-H protons protected from exchange in the molten globule B,C helices in -lactalbumin are protected in the acid molten globule state. 4. NMR of the molten globule state is much simpler than the native molecule. Some pronounced resonance at < 1 ppm in the native form completely disappear in the molten globule. 5. Environment of many side chains is much less rigid in the molten globule vs the normal state (Spin-spin relaxation time diminishes) Motions of methyl groups coincide with the unfolded state while motions of aromatics is intermediate between native and unfolded Why is denaturation so widely studied ? It is thought that denaturation pathways are the opposite of folding pathways. Old idea-the concept of an all or nothing transition to a completely unfolded state is probably wrong. The all or nothing transition is probably native molten globule (movement of side chains, destruction of tight tacking) Then the system goes from molten globule random coil through loss of secondary structure. 21 Facts about Molten Globule States in Proteins 1. Proteins can be transformed into the MG state by low or high pH, by high temperature, by moderate levels of urea or guanidine HCl, and by the influence of LiClO4 and other salts (i.e. under mild denaturing conditions). 2. Proteins can be transformed into the MG state without a change of the environment , simply by small alterations of their chemical structure. Examples: I. Staphylococcal nuclease with 21 C terminal residues removed II. Point mutants of lambda repressor III. BPTI with reduced S-S bonds IV. Alpha-lactalbumins after Ca2+ removal. 22 Example: Urea induced unfolding of carbonic anhydrase B at pH 3 compared with pH 7.5 Folding pathway. Folding starts with the formation of fluctuating embryos of regions with secondary structure (stabilized mainly by h-bonds), followed by collapse of these regions into an intermediate compact structure that is stabilized mainly by hydrophobic interactions. The final structure is driven by van der Waals/ and other specific interactions. Major Problem: Does any of the above happen in cells? 1. Enzymes exist that accelerate cis-trans isomerization in proline residues, and others catalyze S-S bond rearrangement. 2. All cells contains “chaperonins”, which either aid protein folding or prevent protein folding or prevent proteins from associating prematurely with other proteins. These were discovered as heat shock proteins accumulated after cells were subjected to temperature jumps or other stress. Some chaperonins prevent improper folding of membrane proteins-or prevent membrane proteins from aggregating. GroEL is a beautiful molecular machine. The protein to be protected is bound within the central cavity as a molten globule. 23 Chaperonins - In addition to the enzymes mentioned previously that assist with proper folding (e.g., cis-trans isomerase for proline and disulfide bond making enzymes), cells have a class of proteins called chaperonins, which "chaperone" a protein to help keep it properly folded and non-aggregated. Aggregation is a problem for unfolded proteins because the hydrophobic residues, which normally are deep inside of a protein, may be exposed when the protein is released from the ribosome. If they are exposed to hydrophobic residues in other strands, the two strands may associate with each other hydrophobically (to aggregate) instead of folding properly. These proteins were first identified as heat shock proteins , induced by elevated temperatures or other stress. The most thoroughly studied are hsp-70 (70 kD) and hsp-60 (60-kD). The GroEL-ES complex from E. coli is one such chaperone system (hsp-60). It provides a central cavity in which new protein chains can be "incubated" until they have folded properly . 24 An Amazing Molecular Machine 25 The best studied chaperonin: GroEL-ES complex from E. Coli GroEL has two rings each with 7 protein molecules. The center of each ring has the open hole which is accessible to the solvent. Either cavity can be capped with Gro-ES again a seven membered ring of smaller subunits. The insulating property of this molecular machine prevents aggregation or misfolding. The process is ATP-hydrolysis driven Hypothetical Model for chaperonin action in Rubisco folding. Active dimer (top) can be unfolded (e.g. 8M Urea) to give an unfolded polypeptide. The dimer can also be acid-denatured to give a polypeptide that still retains elements of secondary structure. It is suspected that a common intermediate forms from either of these two states on removal of the denaturant. This intermediate is labeled Rubisco I. In the absence of chaperonins, dilution of denatured Rubisco leads to precipitation. If cpn -60 (a chaperonin) is present during dilution, a binary stable complex is formed between it and Rubisco I. When cpn10 and MgATP are added to this complex in the presence of K+ ions, active Rubisco dimers are formed. 27 Rubisco-major protein component of chloroplasts, possibly the most abundant protein in the world. Its function is the key step in CO2 fixation in the Calvin cycle of photosynthesis. 26 + GroE - GroE Suppression by groE complex of aggregation during refolding. Citrate Synthase was denatured, and refolding was initiated by 100-fold dilution of the unfolded protein to the indicated protein concentration. The refolding was measured in the presence and absence of the GroE complex. A 6-fold molar excess of GroE complex over CS was used. 28 Prediction of Secondary and Tertiary Protein Structure Investigators have examined the structures found in proteins and tried to relate them to the individual amino acids. Secondary Structure - Table 6.6 lists the relative probabilities that a particular amino acid will form an -helix, -sheet, and a "turn" in proteins. Note that the top group of amino acids favors -helices, the middle group favors -sheets, and the last group favors turns. The Chou-Fasman rules for predicting secondary structure of a region of a polypeptide sequence are the following: 1. Any segment of 6 residues or more with an -helix probability of over 1.03, and not including proline or phenylalanine, is predicted to be -helix. 2. Any segment of 5 residues or more, with beta -sheet probability greater than 1.05 (except histidine) is predicted to be -sheet. 3. Tetrapeptides with an helix probability less than 0.9 and a turn probability greater than a -sheet probability have a good chance of being turns. The secondary structures observed in the native protein BPTI as predicted .by the Chou-Fasman rules provides exceptionally good agreement between prediction and experiment. 29 Hydrogen exchange of individual backbone amide protons in BPTI followed by 2D NMR methods. The protein was 30 dissolved in D2O and kept at 36 C for the indicated times before spectra were acquired.The resonances from the amide protons that disappear are identified on the last spectrum on which they were apparent, using the one letter code for the amino acids. Prediction of protein structures Rose and Srinivasan assume that protein folding is both local and hierarchichal. Local means that each amino acid’s folding is influenced by other residues nearby in the sequence. Hierarchichal means that folded structures develop from the smallest structural units and work up to more and more complex entities. The program they have developed is called LINUS “Local Independently Nucleated Units of Structure” The program considers groups of three amino acids in a sequence -for example residues 12,13,1 4 in a group of 50. The initial assumption is that this group will (randomly) adopt one of 4 possible structures helix, sheet, turn or loop (other). The program then asks whether this assumed “ministructure” is energetically suited to the six amino acids on either side. The program then moves on to the next (overlapped) set of residues 13, 14, 15 in this case. This random selection and testing is carried out many times for the entire protein. Once this is done, the program analyzes all trials, looking for local groups of amino acids that prefer one of the four conformations 70% of the time. Such groups are held in these conformations while the program repeats the entire process using comparisons of energetic preferences with respect to 12, amino acids on either side of selected groups of three residues, then 18 amino acids and so on up to 48 residues. 31 The results of LINUS for some proteins are shown below: Actual and predicted structures of three domains of intestinal fatty acid binding protein Actual and predicted structures of a helical domain of cytochrome b-562 32 33