Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Polysubstance dependence wikipedia , lookup
Orphan drug wikipedia , lookup
Cannabinoid receptor antagonist wikipedia , lookup
Discovery and development of angiotensin receptor blockers wikipedia , lookup
NK1 receptor antagonist wikipedia , lookup
Compounding wikipedia , lookup
Plateau principle wikipedia , lookup
Psychopharmacology wikipedia , lookup
Nicotinic agonist wikipedia , lookup
Pharmacognosy wikipedia , lookup
Pharmacogenomics wikipedia , lookup
Pharmaceutical industry wikipedia , lookup
Prescription costs wikipedia , lookup
Prescription drug prices in the United States wikipedia , lookup
Theralizumab wikipedia , lookup
Drug discovery wikipedia , lookup
Drug design wikipedia , lookup
Neuropsychopharmacology wikipedia , lookup
Drug interaction wikipedia , lookup
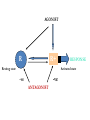
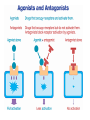
INTRODUCTION to Pharmacology What is pharmacology? • It is the study of the actions, mechanisms, uses, and adverse effects of drugs • A Drug is any natural or synthetic substance that alter the physiologic state of a living organism Drugs are divided into: • Medicinal drugs: These are used in the prevention, treatment and diagnosis of disease • Non-medicinal drugs or social drugs: These are used for purposes other than treatment e.g. illegal mood- altering substances such as cannabis, heroin and cocaine Drugs name and classification Factors that are used to classify drugs: 1. Pharmacotherapeutic action 2. Pharmacologic action 3. Molecular action 4. Chemical nature Definitions • Pharmacodynamics: • It is what the drug do to the body i.e. drug actions on the receptor, tissue, organ or system. • Pharmacokinetics: • It is what the body to the drug i.e. absorption, distribution, metabolism and elimination Pharmacodynamics Targets of drugs • • • • • • Receptors Enzymes Ion channels Carrier molecules Physical Chemical Drug receptors • Highly specific, highly sensitive binding site • Found on the surface of the cell membrane, enzyme molecules on nuclear membranes. • Receptor Specificity – There are a number of specific ligands and a number of associate receptors • Affinity • The extent to which the ligand is capable of binding and remains bound to a receptor • High affinity: The ligand bind well and remains bound for long enough to activate the receptor • Low Affinity: The ligand binds less well and may not remain bound long enough to activate the receptor The receptor intrinsic activity • The extent to which the ligand activates the receptor • High intrinsic activity: the ligand produces a large effect on the post synaptic cell • Low intrinsic activity: the ligand produces a small or inconsistent effect on the post synaptic cell Classes of Ligands • Agonists – High affinity – High intrinsic activity • Antagonist – High affinity – Low intrinsic activity Agonists • An agonist is a drug that binds to a receptor and activates it to give a response • There are 2 types of agonists: 1. Full agonist: is a drug when bound to a receptor produces 100% of the maximum possible biologic response 2. Partial agonist: a drug that produce less than 100% of the maximum possible biologic response no matter how high their concentration AGONIST R R+ Resting state RESPONSE Activated state -ve -ve ANTAGONIST Antagonist • An antagonist is a drug that bind to a receptor and produce no response. • Type of drug antagonism: 1. Receptor antagonists a) Competitive: the antagonists competes with the agonist at the same receptor site of an agonist and binds to it reversibly. By increasing the concentration of drug agonist the antagonism is abolished. E.g. antihistamine competes with histamine at its receptor site b) Non competitive: the antagonist binds irreversibly to a different site on the enzyme e.g. than the agonist, noncompetitive agonists can not overcome by increasing the concentration of the drug agonist. E.g. succinylcholine is irreversible neuromuscular blocker. Continue … 2. Physiological antagonism: e.g. glucagon and insulin 3. Chemical antagonist: is the type of a drug that acts by binding chemically to the compounds and inactivates it. E.g. chelating agents, charcoal (poisoning with iron) 4. Pharmacokinetic antagonist: by increasing the metabolism and excretion of a drug, e.g. enzyme induction to speed metabolism Antagonist Reversible: • Can be unbound from the receptor – Irreversible: • Cannot be unbound from the receptor – Competitive: • Competes with other ligands for binding to the – receptor Non competitive: • Exerts its antagonist effects without competition for – occupancy of the receptor Presynaptic Adrenergic Neurons (Blue balls = NE) Yellow Balls = Beta Blocker Dose- response curve • The response to increasing doses of an agonist or antagonist are plotted graphically. • It can measure the following: • Efficacy: is the maximum effect (Emax) which can be obtained from a large dose of the drug. • Potency: is the amount (dose) of drug or concentration in the plasma that can produce a similar effect by a dose of another drug • EC50 (Effective concentration): is the concentration that produce 50% of the maximum effect. • ED50 (Effective dose): is the dose that produces the response of 50% of subjects • LD50 (Lethal dose): is the dose that cause death in 50% of subjects • Non competitive antagonist will bind irreversibly with the receptor leading to decrease in number of receptors available for binding with the agonist • This will cause the maximum effect of the agonist to be less than when it is used alone or with competitive antagonist Therapeutic index • Is the ratio between the average maximum tolerated dose LD50 and the average minimal effective dose ED50 TI= LD50/ ED50 • It indicates the margin of safety in use of a drug • The higher the TI the safer the drug Type of receptor proteins A. ligand gated ion cannels: • Ionotropic receptors • They are membrane receptors that compose an ion channel with ligand binding site (receptor) in the extracellular domain that stimulate the opening of the channel. • It increases Na, K permeability and possibly generates an action potential. • Typically are these the receptors which fast neurotransmitters acts (milliseconds). • E.g. nicotinic Ach receptor, GABA A receptor B. G-protein coupled receptors • Know as metabotropic receptor, they are membrane receptors • They either stimulate an enzyme or a channel • The receptor is coupled with G protein • G-proteins comprises 3 subunits; α, β, γ . The α subunit consists of GTPase, which catalyzes the conversion of GTP to GDP and break down the G-proteins releasing the α subunit • The α-GTP diffuse in the membrane and associate with various enzymes causing activation or inactivation. • This process is terminated when the α subunit is recombined with the β, γ subunits Na+ Neuromuscular Blocking Drugs Competitive Tubocurarine Gallamine Pancuronium Vecuronium Atracurium Rocuronium Depolarizing Suxamethonium ACh Na+ α ACh δ γ α β • Classes of G- protein : Gi, Gs, Gq. Gs & Gi either stimulate or inhibit enzyme Adenylate cyclase. Gq activates phospholipase C (PLC) • Example: cholera toxin acts by Gs pertusis toxin acts by Gi • Targets of G- proteins: 1- adenylate cyclase (cAMP) 2- Phospholipase C PLC(IP and DAG) 3- Ion channels (Ca+2 and K+) Cholinergic nerve terminal Neuromuscular Blocking Drugs Action Potential Arrives Acetyl CoA + Choline Cholineacetyl Transferase ACh Ca++ ACh ACh Potentiate Transmission Pyridostigmine Neostigmine Distigmine edrophonium Synaptic Cleft ACh ACh ACh ACh Agents that reduce ACh release Vesamicol ACh ACh Hemicholinium Botulinum toxin Aminoglycosides Mg2+, Ca2+ ions ACh ACh ACh ACh Competitive Tubocurarine Gallamine Pancuronium Vecuronium Atracurium Rocuronium Depolarizing Suxamethonium Na+ Acetate ion + Choline Acetylcholinesterase inhhbitors G P C R Post-Synaptic membrane Receptors) G-protein coupled receptors • Membrane bound receptors which are bound to effector system through G-proteins • These are hetero trimeric molecules having 3 subunits alpha, beta, and gamma • Based on alpha subunits they are further classified into 3 main subtypes Gs, Gi, and Gq C- Kinase- linked and related receptor: • They mediate the action of many different mediators including growth factors, cytokines, and hormones (insulin) • They have a common structure, a large extracellular (ligandbinding) domain connected by an α- helix to an intracellular domain (effector) D- Nuclear receptors • These are ligand for: steroid hormones, thyroid hormones, vt.D and retinoic acid as well as lipidlowering drugs and antidiabetics • From its name it is found in the nucleus (intracellular) so the ligand must enter the cell. • The compounds are usually lipophilic so they readily cross the lipid-bilayer. • They are important for gene expression, alter protein synthesis and drug metabolizing enzymes. • They are the slowest Other GATING mechanisms: 1- voltage- gating channels: • They open when the membrane is depolarized (i.e. excitation) in the activation phase • It is short lasting • They are either Na+, K+, Ca+2 voltage- gated channel 2- calcium releasing channels: • They lie on the membrane of the endoplasmic reticulum Functions of receptors • To propagate signals from outside to inside • To amplify signals • To integrate various extracellular and intracellular regulatory signals • To adapt to long term changes in maintaining homeostasis Undesirable responses to drug therapy • Toxic effect : – When too much drug has collected in the patient. – It may be due to: – an acute high dose of a drug – chronic buildup over time –or increased sensitivity to the standard dose of a drug. • Drug allergy (hypersensitivity): – The body sees the drug as an antigen and an immune response is established against the drug. – This may be an immediate response or delayed • Adverse reaction– undesirable drug effect. • Side effect– This is similar to the term adverse reaction. – It is an unwanted but expected responses to a drug. Adverse Drug Reaction • Frequency of adverse drug reaction • ADRs are common. • About 2-6% of hospital admissions are for ADRs. Types of ADR’s • A: – Augmented – dose related – Related to pharmacology (toxic effect or side effect--for example, digoxin toxicity) • B – Bizarre – non-dose related, – Unrelated to pharmacology (idiosyncratic for example, malignant hyperthermia, or immunological--for example, penicillin rash) • C: – Continuous or chronic – Dose and time related, – Related to cumulative drug use--for or chronic example, NSAID induced renal failure • D: – Delayed,, – Delayed effect – Can be seen only some time after use of drug--for example, ??? • E: – End of use – Withdrawal – Related to discontinuation that is too abrupt-for example, addisonian crisis after steroid withdrawal Factors affecting a patient’s response to a drug • Age. – The very young – The elderly. • Body weight. • Pregnancy and lactation. • Nutritional status. • Food-drug interactions. For example, orange juice (vitamin C) will enhance the absorption of iron sulphate, but dairy products reduce the absorption of tetracycline.. • Disease processes. Includes: – Changes in gut motility e.g. diarrhoea – Loss of absorptive surface in the small intestine, as occurs in Crohn’s disease will affect absorption. – Hepatic disease – Renal disease – Circulatory diseases – Mental and emotional factors. – Genetic and ethnic factors. PHARMACOKINETICS Translocation of drug molecules Drug molecule move around the body in 2 ways: 1. in blood stream (bulk transfer), the chemical nature of a drug is of no importance to its transfer. 2. molecule by molecule, over short distance (diffusional transfer), the chemical nature of a drug is an important factor especially its ability to cross hydrophobic barriers. The rate of diffusion depends also on its molecular size, large molecules tend to diffuse more slowly than smaller ones. Movement of drug molecule across cell barriers Cell membrane form the barrier between aqueous compartments of the body epithelial barrier such as GI mucosa or renal tubules, consist of a layer of cells tightly connected to each other Vascular epithelium: Gabs between endothelial cells are packed with a loose matrix of proteins that acts as filters, retaining large molecules and letting smaller ones through. In some organs, especially CNS and placenta, there are tight junctions between the cells and the endothelium is an impermeable layer. These features prevent potentially harmful molecules from leaking from the blood to these organs. In other organs (e.g. liver and spleen), the endothelium is discontinuous, allowing free passage between cells. There are 4 main ways by which molecules cross the cell membrane: 1) by diffusion directly through lipids 2) by diffusion through aqueous pores formed by special aquaprotiens that traverse the lipids 3) by combination with transmembrane carrier protein that binds a molecule on one side of the membrane then changes configuration and release it to the other side. 4) by pinocytosis PH ionization of weak acids and weak bases, the Handersons-Hasselbakh equation The electrical charge of an ionized molecule attracts water dipoles and results in polar, relatively water soluble compound. Since lipid diffusion depends on relatively high lipid solubility, ionization of drugs may markedly reduce their ability to permeate membranes. Weak acid is best defined as a neutral molecule that can reversibly dissociate into an anion and a proton. The protonated form is neutral and more lipid soluble HA↔H+ +AWeak base is defined as neutral molecule that can form a cation by combining, with a proton, the un-protonated form is the neutral form B H+↔H+ +B The HHE relates the ratio of protonated to unprotonated weak acids and weak bases to the molecules PKa and PH of the medium as follows: Log 10 protonated = PKa _ PH unprotonated For bases: PKa = PH + Log 10 [BH+] [B] For acids: PKa = PH + Log 10 [AH] [A-] PKa: (ionization constant), is PH at which 50% of the drug is ionized. Because the uncharged form is the more lipid soluble, more of weak acids will be lipid soluble in acidic medium and more weak bases will be lipid soluble in alkaline medium. Ionization increases renal clearance of drugs • Both ionized and non-ionized forms of a drug are filtered • Only free unbound drug is filtered • Only non-ionized forms undergo secretion and active or passive reabsorption • Ionized forms of drugs are trapped in the filtrate Fick’s law of diffusion The passive flux of molecules down a concentration gradient is given by fick’s law Flux (molecule per unit time)= (C1-C2) × area × permeability coefficient × Thickness C1: the higher concentration C2: the lower concentration Area: is the area across which the diffusion is occurring Permeability coefficient: is a measure of the mobility of the drug molecule in the medium of the diffusion bath Thickness: is the thickness (length) of the diffusion bath DRUG DISPOSION DIVIDED INTO: – Absorption – Distribution – Metabolism – Excretion Drug forms 1. 2. 3. 4. 5. Tablets Capsules Solution: for injections, either IM, IV, SC Solution to drink Preparation to apply to skin or mucus membranes 6. Vaginal tablets 7. Rectal preparation Routes of drug administration • • • • • • Oral Sublingual Rectal Application to other epithelial surfaces (cornea, skin, vagina and nasal mucosa) Injections (SC, IM, IV, intra-thecal) Inhalation ORAL ROUTE • Taken by mouth • Drugs are absorbed by: • Passive Diffusion: o Determined by the lipophilcity of the drug compound o Ionized drugs are poorly absorbed (strong base or acid) • Active Diffusion: o By carrier proteins o Example: ca+2 is carried by V.t-D dependent carrier system Factors affecting GI absorption 1- Altered gastric motility e.g. diarrhea 2- Splanchnic blood flow (it decreases in shock) 3- Particle size and formulation 4- Physiochemical Factors: i.e. ions that affect the absorption of some drugs. e.g. Tetracycline: it binds to ca and ca containing food SUBLINGUAL ROUTE • It is useful when rapid response is required specially if the drug is unstable or has an increase first pass. • The drug by pass the portal circulation • Example: Glyceryl trinitrate RECTAL ROUTE: • Used for either local effect (anti-inflammatory drugs used for ulcerative colitis) or to produce a systemic effect like Diazepam in children who present with epilepsy. Other local applications: • • • • Cutaneous administration Nasal spray (ADH, GnRH, Calcitonin) Eye drops Inhalation (Ipratropium, salbutamol) PARENTRAL ADMINSTRATION Intravenous injection: • Fast and most effective • Directly enter the venous circulation then to the right side of the heart then to the lungs the to the systemic circulation. Intramuscular injection: • They fastest than the oral route. • The rate of absorption depends on the site of inj. And the blood flow. • Increase the blood flow: exercise, rubbing, hot bath • decrease the blood flow: cooling Intra- thecal injection: • The injection in the subarachnoid space Example: • Methotrexate: given for treatment of leukemia • Regional anesthesia BIOAVAILABILITY • Defined as the proportion of drug conc that reach the systemic circulation following administration. • This is measured by the area under the curve AUC AUC= A / Ke • A: the point or concentration where the drug reach max plasma concentrated • Ke: elimination rate constant (0.7/ t½) f = AUC PO/AUC IV A DRUG CONCENTRATION DRUG CONCENTRATION A Ke AUC IV 0 TIME AUC ORAL 0 Ke TIME Ke= 0.7/ t½ • Ke: the time were the concentration of the drug in plasma drop by 50% (elimination constant) • IV doses have 100% bioavailability, f = 1 Factors affecting Bioavailability A- Extent of absorption: • Usually any drug taken by oral administration is incompletely absorbed B- First- pass metabolism: • After absorption of a drug it goes to the liver through portal circulation were it is metabolized to active or inactive compounds (can occur in the gut). • Some of these compounds are excreted in bile. VOLUME OF DISTRIBUTION • Vd: relates to the amount of drug in the body to the concentration of the drug in blood or plasma Vd= amount of drug in body C • It is affected by protein binding. • Only unbound drug (free fraction) exerts pharmacolological effects Special Barriers to Distribution Placental • Most drugs cross the placental barrier, but fetal blood level is usually lower than maternal Blood-Brain • Permeable to lipid soluble or very small drug molecules Redistribution • Lipid-soluble drugs redistribute into fat tissues prior to elimination - repeated doses cause saturation – may prolong duration of action • The higher the Vd, the lower the plasma concentration and vice versa • Vd is low when a high % of drug is bound to plasma proteins Hepatic Extraction ratio (ER) • It is affected by: rate of hepatic clearance of the drug and hepatic blood flow ER= CL liver/ Q • CL liver: liver clearance of the drug • Q: hepatic blood flow Systemic Bioavailability (F) F= f(1-ER) • f: the extent of absorption of the drug Example: A drug like morphine if taken orally almost completely absorbed f=1 hepatic ER=0.67 so F=(1-ER)=1-0.67=0.33 F= 0.33×100=33% DRUG ELIMINATION • It involves the following: 1- Drug metabolism: which include enzymatic conversion of one chemical entity to another 2- Drug excretion: includes the elimination of drugs either unchanged or metabolized • It occurs through the kidney, hepato-billiary and lung systems Drug Metabolism • Lipophilic compound are not excreted by the kidney and they need to become more soluble or polar to be excreted. • Metabolism occurs mainly in the liver (CYP450 system) • Metabolism may result in formation of active metabolites (diazepam – nordiazepam) • Prodrugs lack activity until they undergo bioactivation (clorazepate – nordiazepam) Metabolism involves two phases: Phase -1 reaction: (catabolic) • it includes: oxidation, reduction, and hydrolysis reaction. • It is called “the microsomal mixed function oxidase system”. • Major phase 1 enzymes – localized in smooth ER of liver, GI tract, lungs, and kidneys • It includes two enzymes: 1) NADPH cytochrome reductase 2) CYP450 system • Require O2 and NADPH • Multiple CYP families vary by substrate specificity and sensitivity to inhibitors & inducing agents CYP3A4 • Major isoform with wide substrate range • Inhibited by cimetidine, macrolides, azoles & ethanol (acute) • Induced by carbamazepine, phenobarbital, phenytoin, rifampicin, & ethanol (chronic) CYP2D6 • Genotypic variations in hydroxylation (fast / slow) • Substrates include codeine, debrisoquin & metoprolol • Inhibited by haloperidol & quinidine; not inducible Other Phase 1 metabolism Non-microsomal oxidations • Monoamine oxidases metabolize NE, 5HT, and tyramine • Alcohols metabolized via alcohol dehydrogenase (ADH) to aldehydes then aldehyde dehydrogenoase (inhibited by disulfram) Phase -2 reaction: • It include conjugation with other groups to make it more soluble. • Groups used in conjugation: glucoronyl, sulfate, methyl, acetyl, glycyl glutathione • This phase can occur in the kidneys • Acetylation is genetically determined. Fast acetylators and slow acetylators (develop SLE like syndrome when given INH, hydralazine, procainamid or INH • Transferases: usually inactivate drugs, but may activate (e.g. morphine, minoxidil). May follow a phase I hydroxylation, but also occur directly • Glucuronidation – inducible; reduced activity in neonate RENAL EXCRETION There are 3 main process for Renal excretion of a drug: 1- Glumerular filtration rate: (GFR) • This depends on the molecular weight of the drug and the extents of binding to plasma proteins. 2- Tubular secretion: • Here drug molecules are transferred by two independent and non-selective carrier systems i.e. Transport of acidic compounds or basic compounds • They transport drug molecules against conc. gradient so can reduce the plasma conc. of the drug to zero. • E.g. penicillin 3- Diffusion across the renal tubules: • Renal tubes can be freely permeable (the drug concentration in the plasma and in the renal tube is equal) DRUG ELIMINATION (CLEARANCE) • Defined as the volume of plasma containing the amount of substance that is removed by the kidney in unite time CL= Cu Vu Cp CL: Clearance Cu: Urine Concentration Cp: Plasma Concentration Vu: Volume of Urine CLEARANCE • • • • Equals rate of elimination divided by plasma level Constant for 1st order elimination Total body clearance CL = CLR + CLER (extra renal) With no secretion or reabsorption renal clearance is the same as glomerular filtration rate, CLR = GFR • If drug is protein bound then CLR = GFR x free fraction There are two ways for drug elimination: 1- First Order Kinetic 2- Zero Order Kinetic First Order Kinetic (un- saturable) • Defined as the amount of drug removed is direct proportion to its concentration in plasma. ZERO- ORDER KINETICS (SATURABLE) • Her drugs are removed at a constant rate regardless the plasma concentration levels because it is an enzyme dependant process so it has limited capacity. Example: • Ethanol • Phenytoin • Salicylates Blood Alcohol concentration t½ 10.9 7.6 4.3 TIME Dose Administration Alcohol is eliminated at a rate of 4mmol/l regard less the plasma concentration t½ HALF LIFE OF A DRUG • Is the time required by the body to eliminate 50% of the drug concentration t½= Vd x 0.7 CL It is important to indicate the time required to attain 50% of the steady state • This helps in the setting up of a dosage regime which produces: – stable plasma drug concentrations – keeps the level of drug below toxic levels but above the minimum effective level • Loading Dose – This is given when an effective plasma level of drug must be reached quickly. – This requires a dose of the drug which is larger than is normally given. – This dose is given as a one off. • Maintenance dose: – This is the dose given when the required plasma level of drug has been reached. – It is the normal recommended dose. – This is then continued at regular intervals to maintain a stable plasma level .