Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Oligonucleotide synthesis wikipedia , lookup
Biochemical cascade wikipedia , lookup
Nicotinamide adenine dinucleotide wikipedia , lookup
Oxidative phosphorylation wikipedia , lookup
Metalloprotein wikipedia , lookup
Metabolic network modelling wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Ribosomally synthesized and post-translationally modified peptides wikipedia , lookup
Basal metabolic rate wikipedia , lookup
Mitogen-activated protein kinase wikipedia , lookup
Paracrine signalling wikipedia , lookup
Nucleic acid analogue wikipedia , lookup
Point mutation wikipedia , lookup
Fatty acid synthesis wikipedia , lookup
Fatty acid metabolism wikipedia , lookup
Peptide synthesis wikipedia , lookup
Citric acid cycle wikipedia , lookup
Protein structure prediction wikipedia , lookup
Proteolysis wikipedia , lookup
Genetic code wikipedia , lookup
Biochemistry wikipedia , lookup
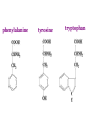
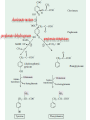
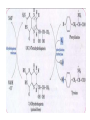
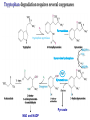
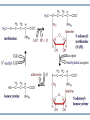
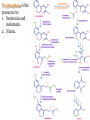
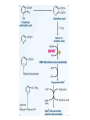
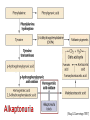
AROMATIC AMINO ACID Aromatic amino acid contains aromatic ring. They are: Phenylalanine(Benzene ring or phenyl). Tyrosine(phenol). Tryptophan(indole). Phenylalanine and Tryptophan are essential amino acids. Tyrosine can synthesis from Phenylalanine. Partially glucogenic and partially ketogenic. Only plants and microorganisms are able to synthesize aromatic amino acids. phenylalanine tyrosine tryptophan Biosynthesis of aromatic amino acids: Biosynthesis of aromatic amino acids starts with a common pathway, the Shikimate pathway. The biosynthesis begins with Phosphoenolpyruvate and Erythrose-4- phosphate to form Shikimate. Shikimate then goes on to form the branch point intermediate Chorismate. Chorismate can be converted into anthranilate (L-Trp) or prephenate (L-Phe and L-Tyr). Shikimate pathway : 2-keto-3-deoxy-D-arabinoheptulosonate-7- phosphate synthase. : dehydroquinate synthase. : 3-dehydroquinate dehydrogenase, : shikimate dehydrogenase,: shikimate kinase, : 3-enolpyruvylshikimate-5-phosphate synthase, and : chorismate synthase. Tryptophan synthesis Tryptophan is synthesized from chorismate in a 5-step process. Chorismate requires an amino group from the side chain of glutamine and releases pyruvate to form anthranilate. Anthranilate then undergoes condensation with phosphoribosyl pyrophosphate (PRPP), an activated form of ribose phosphate. The C-1 atom of ribose 5-phosphate becomes bonded to the nitrogen atom of anthranilate in a reaction that is driven by the hydrolysis of pyrophosphate. The ribose moiety of ribosylanthranilate undergoes rearrangement to yield enol-1-o-carboxylphenylamino-1deoxyribulose-5-phosphate. This intermediate is dehydrated and then decarboxylated to indole-3- glycerol phosphate, which reacts with serine to form tryptophan. : anthranilate synthase, : anthranilate phosphoribosyl transferase, : N-(5’-phosphoribosyl)- anthranilate isomerase, : indole-3-glycerol phosphate synthase, and : tryptophan synthase. Tyrosine and phenylalanine synthesis A mutase converts chorismate into prephenate, the immediate precursor of the aromatic ring of tyrosine and phenylalanine. Prephenate is oxidatively decarboxylated to phydroxyphenylpyruvate. Alternatively, dehydration followed by decarboxylation yields phenyl pyruvate. These α-keto acids are then transaminated, with glutamate as amino group donor, to form tyrosine and phenylalanine, respectively. chorismate mutase prephenate dehydrogenase prephenate dehydratase Tyrosine can also be made by animals directly from phenylalanine via hydroxylation at C-4 of the phenyl group by phenyl hydroxylase, which also participates in the degradation of phenylalanine. Hydroxylation of phenylalanine to form tyrosine involves the reductant tetrahydrobiopterin. Dihydrobiopterin is reduced to tetrahydrobiopterin by electron transfer from NADH. Thus NADH is secondarily the e- donor for conversion of phenylalanine to tyrosine Degradation of Aromatic Amino Acids (1) Transaminase. (2)p-hydroxy-phenylpyruvate dioxygenase (vitamin Cdependent). (3)homogentisate dioxygenase . (4)4-Maleylacetoacetate isomerase . (5)fumarylacetoacetase. Tryptophan degradation requires several oxygenases Formamidase tryptophan pyrrolase kynurenine hydroxylase NADPH + H+ NADP+ PLP Kynureninase Pyruvate NAD and NADP Amino Acids as Metabolic Precursors Tyrosine is the precursor to several important molecules in metabolic signaling and neurotransmission, including epinephrine and dopamine. Tyrosine is oxidized by the enzyme tyrosine hydroxylase in a reaction requiring the enzyme cofactor tetrahydrobiopterin to form dihydroxyphenylalanine (L-DOPA), a metabolic precursor to dopamine Tyrosine is also the precursor to pigment molecules called melanins that are produced from dopaquinone. The two primary melanins are eumelanins, which are dark pigments having a brown or black color, and pheomelanins that have red or yellow color. The yellow color of pheomelanin pigments comes from the sulfur in cysteine that is combined with dopaquinone. Thyroid hormones - Thyroxine (tetraiodothyronine) & triiodothyronine - are synthesized from the tyrosine residues of the protein thyroglobulin & activated iodine. Iodination of tyrosine ring occurs to produce mono & diiodotyrosine from which triiodothyronine (T3) & thyroxine (T4) are synthesized. The protein thyroglobulin undergoes proteolytic breakdown to release the free hormones - T3 & T4. Tryptophan, is the precursor to: 1. Serotonin and melatonin. 2. Niacin. INBORN ERRORS OF AMINO ACIDS METABOLISM: Alcaptonuria - inherited disorder of the tyrosine metabolism caused by the absence of homogentisate oxidase. homogentisic acid is accumulated and excreted in the urine turns a black color upon exposure to air In children: urine in diaper may darken In adults: darkening of the ear dark spots on the on the sclera and cornea arthritis phenylketonuria (PKU) A genetic defect in the gene encoding phenylalanine hydroxylase is responsible for the metabolic disease phenylketonuria (PKU). Defect in myelination of nerves The brain weight is below normal. Mental and physical retardations. The life expectancy is drastically shortened. Diagnostic criteria: phenylalanine level in the blood FeCl3 test DNA probes (prenatal) Treatment: consists of limiting phenylalanine intake to levels barely adequate to support growth. Tyrosine, an essential nutrient for individuals with phenylketonuria, must be supplied in the diet. The clinical symptoms of PKU are caused by the accumulation of phenylalanine in the blood that is 30-50 times higher than normal. This high level of phenylalanine leads to the production of phenylalanine metabolites such as phenylpyruvate, phenylacetate and phenyllactate, all of which are associated with the observed neurological and developmental problems NutraSweet contains a phenylalanine derivative Phenylketonuriacs also have to be careful to avoid processed foods and beverages containing the food additive aspartame (aspartyl-phenylalanine methyl ester). Albinism – genetically determined lack or deficit of enzyme Tyrosinase Phenylalanine Tyroxine Tyrosine Tyrosinase in melanocytes oxidases tyrosine to DOPA and DOPA-chinone. Melanin tyrosinase DOPA Symptoms of albinism: inhibition of production or lack of melanin in skin, hair, eyes increased sensitivity to sunlight increased risk of skin cancer development sun burns photophobia decrease of vision acuity strabismus, nystagmus Dopamine Norepinephrine Epinephrine Tyrosinemia: occur in several forms. They may be caused by a deficit of enzymes which catalyze either the transamination of tyrosine (II), or oxidation of phydroxyphenylpyruvate (III) and hydrolysis of fumarylacetoacetate (I). A low-tyrosine diet may be very useful. Plasma levels of tyrosine are elevated, and large amounts of tyrosine, phydroxyphenylpyruvate, –lactate, and –acetate are excreted into the urine (tyrosyluria). Thank you