Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Microbial metabolism wikipedia , lookup
Fatty acid synthesis wikipedia , lookup
Gaseous signaling molecules wikipedia , lookup
Pharmacometabolomics wikipedia , lookup
Catalytic triad wikipedia , lookup
Basal metabolic rate wikipedia , lookup
Oxidative phosphorylation wikipedia , lookup
Fatty acid metabolism wikipedia , lookup
Citric acid cycle wikipedia , lookup
Enzyme inhibitor wikipedia , lookup
Evolution of metal ions in biological systems wikipedia , lookup
Genetic code wikipedia , lookup
Metalloprotein wikipedia , lookup
Biosynthesis wikipedia , lookup
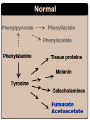
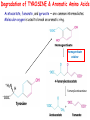
INBORN ERRORS OF AMINO ACIDS METABOLISM TYROSINE METABOLISM Tyrosine Tyrosine is formed from phenylalanine by phenyl alanine hydroxylase. During the reaction, BH4 is oxidized to dihydro biopterin (BH2). Tyrosine, like cysteine, is formed from an essential amino acid and is, therefore, non essential only in the presence of adequate dietary phenylalanine. Degradation of Phenylalanine to Tyrosine PA hydro-xylase tetrahydrobiopterin +O2 The reaction requires molecular oxygen and the coenzyme tetra hydrobiopterin (BH4), which can be synthesized from guanosine triphosphate (GTP) by the body. One atom of molecular oxygen becomes the hydroxyl group of tyrosine, and the other atom is reduced to water. During the reaction, BH4 is oxidized to dihydro - biopterin (BH2). BH4 is regenerated from BH2 by NADHrequiring dihydro pteridine reductase. Degradation of TYROSINE & Aromatic Amino Acids Acetoacetate, fumarate, and pyruvate — are common intermediates. Molecular oxygen is used to break an aromatic ring. homogentisate oxidase fumarylacetoacetase INBORN ERRORS OF AMINO ACIDS METABOLISM Alcaptonuria - inherited disorder of the tyrosine metabolism caused by the absence of homogentisate oxidase. homogentisate is accumulated and excreted in the urine turns a black color upon exposure to air In children: urine in diaper may darken In adults: darkening of the ear dark spots on the on the sclera and cornea arthritis Alcaptonuria Accumulation of oxidized homogentisic acid pigment in connective tissue (ochronosis) Aortic valve stenosis in alcaptonuria Arthritis of the spine is a complication of alkaptonuria ochronosis Phenylalanine Albinism – genetically determined lack or deficit of enzyme tyrosinase Tyrosine DOPA Tyroxine tyrosinase Dopamine Tyrosinase in melanocytes oxidases tyrosine to DOPA and DOPA-chinone Norepinephrine Epinephrine Melanin Symptoms of albinism: • inhibition of production or lack of melanin in skin, hair, eyes • increased sensitivity to sunlight • increased risk of skin cancer development • sun burns • photophobia • decrease of vision acuity • strabismus, nystagmus Tyrosinemia is an extremely rare but treatable hereditary disorder. When the body cannot break down tyrosine, high levels build up in the blood and form a toxic substance (known as succinylacetone) in the liver, kidneys, and central nervous system. This means that if tyrosinemia isn't treated, it may cause liver and kidney damage and brain-related problems, such as problems with learning. There are three types of tyrosinemia. The type a child has depends on which enzyme they are lacking. Children with tyrosinemia type 1 (HT-1) are deficient in an enzyme called fumarylacetoacetate hydrolase. If not recognized and treated right away, the condition could be fatal for a child at an early age. However, with treatment, tyrosine levels in the blood can be managed. Dietary restriction of tyrosine and phenylalanine in the treatment of hereditary tyrosinemia type 1 (HT-1). A deficiency of the enzyme fumarylacetoacetase leads to an accumulation of the novel and extremely toxic compound succinylacetone Tyrosinaemia type 1 A deficiency of the enzyme fumarylacetoacetase leads to an accumulation of the novel and extremely toxic compound succinylacetone . This is detected by urine organic acid analysis. Tyrosine concentrations in body fluids are also elevated but can be variable. Affected children typically present with severe liver disease and renal Fanconi syndrome. The drug nitisinone inhibits an enzyme higher in the pathway, 4 hydroxyphenylpyruvate dioxygenase. This prevents the synthesis of succinylacetone and dramatically improves prognosis. Patients continue to need dietary therapy to prevent tyrosine accumulation.