Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Cell-penetrating peptide wikipedia , lookup
Silencer (genetics) wikipedia , lookup
Gel electrophoresis of nucleic acids wikipedia , lookup
Nucleic acid analogue wikipedia , lookup
Non-coding DNA wikipedia , lookup
Community fingerprinting wikipedia , lookup
Molecular evolution wikipedia , lookup
List of types of proteins wikipedia , lookup
DNA supercoil wikipedia , lookup
Genomic library wikipedia , lookup
Deoxyribozyme wikipedia , lookup
Molecular cloning wikipedia , lookup
DNA vaccination wikipedia , lookup
Cre-Lox recombination wikipedia , lookup
Vectors in gene therapy wikipedia , lookup
Genetic engineering wikipedia , lookup
DNA Isolation and Genetic Transformation
Objectives
1.
Isolate DNA from cells of plant tissue
(onion).
2.
Use a bacterial plasmid to insert a gene
into a bacterium (transformation).
Introduction
In one sense, people have been manipulating the genotypes of other organisms ever
since they began the domestication of animals and plants. Up until very recently,
however, these manipulations were purely
by selection. That is, we chose plants and
animals that had desirable characteristics,
and we encouraged them to reproduce. By
careful selection, it was possible to develop
populations that differed dramatically from
their wild ancestors, such as corn and cows.
Selective breeding is limited, however, because it does not directly manipulate or
"engineer" the genetic makeup of organisms.
Over the past few decades, however, biologists have developed methods for transferring genes from one organism to another.
An important step took place in 1928, when
Franklin Griffith was experimenting with
two strains of the bacterium Streptococcus
pneumoniae. One of these strains killed
mice if it was injected into them, while the
other strain was harmless. If Griffith killed
the harmful bacteria with heat, the dead
bacteria were harmless when injected. But,
if he mixed live bacteria of the harmless
strain, with heat-killed bacteria of the harmful strain, the injected mice died. Somehow, the dead bacteria transformed the live
ones, and whatever factor was involved was
not destroyed easily by heat. Griffith called
this phenomenon transformation.
We know now that Griffith's transforming
factor was DNA. Moderate heat denatures
proteins, without destroying DNA. Grif-
page 66
fith's bacteria were able to recover functional (and deadly) genes from the debris of
their heat-killed brethren. As biologists
learned more about the function of the
DNA, and the universality of the genetic
code and machinery, they realized that it
would also be possible to transfer genes
among unrelated organisms. In 1980, biologists first succeeded in placing a human
gene into bacteria, which then manufactured
a human antiviral protein known as interferon. From these early efforts, genetic engineering has grown to become a multibillion dollar growth industry, generally
known as biotechnology, and come to be a
critically important part of medicine, industry, and agriculture.
In lab today we will perform two procedures that will give you a feeling for the
sorts of manipulations involved in working
with DNA and placing genes into cells. The
first of these procedures is the isolation of
DNA from plant tissue. The second is a
bacterial transformation, which is similar in
some ways to the pioneering work done by
Griffith.
1- Isolation of DNA from onion
We could extract DNA from any living tissue, but it is convenient to use a plant, because it won't object when we put it into a
blender. Several obstacles must be dealt
with, however. The DNA is within the nuclear membrane, the cell membrane, and the
cell walls. The cells themselves are massed
into compact tissues. We will homogenize
an onion in a blender, in order to disperse
the cells and mechanically break the cell
walls. Knowing what you do about onions,
you don't want to get too close to this operation! (Incidentally, the "tear gas" that onions release when cut is called synpropanethial-S-oxide. It is produced when
enzymes called allinases are released from
the cells and act on compounds called
amino acid sulfoxides. This reaction proba-
DNA Isolation and Genetic Transformation
bly protects the onion from being eaten by
some organisms).
We will homogenize the onion in an
"extraction solution" that contains the laundry liquid "Woolite" and NaCl. Woolite
contains detergents that will dissolve the
cell membranes, and also proteolytic enzymes that will break down proteins. These
proteolytic enzymes will help to free the
DNA from histone proteins and also help to
inactivate other lytic enzymes that would
break up the DNA. Heating at 60C will further aid these steps- many proteins denature
at 60 degrees Celsius, while DNA is stable
up to 80 C. The salt will help to stabilize
and solubilize the DNA by providing
cations to interact with the acid anions of
the DNA.
After the DNA is in solution, the trick is to
separate it from all the other cellular debris
in the homogenate. It turns out that this is
fairly easy to do. We will create a layer of
another solvent (ethanol) on top of the extraction solution. The DNA is not soluble
in ethanol, and it will precipitate (come out
of solution) at the interface between the
ethanol and the extraction solution. The
strands of DNA are long enough to wind up
on a glass or plastic rod.
Procedure
1. The TA will puree 1 medium onion in
100 ml of extraction solution, and distribute 30 ml of homogenate into a 50
ml centrifuge tube for each group.
2. Quickly cap the tube of homogenate
and place it the at 60 C water bath for
15 minutes.
3. Remove the tube from the water bath
and place it on ice for at least 5 minutes.
4. Use a coffee filter to collect 6 ml of filtrate in the plastic tube provided.
page 67
5. Slowly pipette 9 ml of ice-cold 95%
ethanol down the side of the tube. Be
careful- you do not want the ethanol to
mix with the filtrate- you want it to
form a layer on top.
6. Allow the tube to sit for undisturbed for
2-3 minutes. After this time you should
be able to see strands of DNA precipitating at the interface of the two layers.
7. Use the glass rods provided to "spool"
the DNA strands by twisting the rod
(not by stirring).
2– Transformation
Remember that a gene is the DNA that provides the instructions for making a particular protein.
The expression of genes
(transcription and translation) results in the
phenotypic characteristics of an organism.
"Phenotype" can mean any physical or
physiological characteristic. Genetic transformation means change in the phenotype
that is caused by acquiring a new gene or
genes.
In this procedure, you will genetically transform a bacterium called Escherichia coli (E.
coli for short). This bacterium is the "white
rat" of microbiology, and it has been studied
intensively by biologists. It is an ideal subject for biotechnology applications, because
it grows very rapidly in culture (dividing
every 20 minutes in ideal conditions).
The phenotypic characteristic that we will
alter is the ability of the bacteria to grow in
the presence of an antibiotic called ampicillin. Antibiotics, by definition, are drugs
that kill bacteria or prevent their growth.
Normally, E. coli cannot grow if ampicillin
is present. However, we will change that by
giving the cells a gene for ampicillin resistance- it codes for an enzyme that breaks
DNA Isolation and Genetic Transformation
down ampicillin. Bacterial DNA is usually
a single continuous loop, unlike the linear
DNA of eukaryotic chromosomes. In addition to the big chromosome, bacteria may
contain, replicate, and express other smaller
loops of DNA, called plasmids. These plasmids can be thought of as little extra chromosomes, carrying a few genes that are not
necessarily essential for the cell.
In some ways, plasmids are similar to viruses, in that they can sometimes be passed
from one bacterial cell to another. This
transfer is fairly rare on a "per bacterium"
basis, and only a small percentage of the
cells will take up plasmids in our experiment. However, with millions of bacteria in
a drop of culture, the odds of some of them
taking up plasmids become pretty good.
We can also facilitate the entry of plasmids
into the cells by various treatments. The
ability of a cell to take up a plasmid is
called "competence".
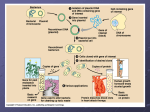
Plasmid pUC18
The DNA that we will use to transform E.
coli is a plasmid called pUC18. Plasmid
pUC18 is a small loop of DNA, containing
2686 base-pairs, and it includes only a couple of genes. Other genes can be spliced
into it, so that it has been used extensively
as a way to get genes into E. coli and
"clone" them.
After a cell takes up a plasmid, it replicates
it, and each time the transformed cell divides, it passes copies of the plasmid on to
its daughter cells. Remember that bacteria
reproduce rapidly, so that any cell can have
thousands of progeny in a matter of hours.
Any cell that takes up the plasmid can rapidly produce a colony of cells that all have
the plasmid.
One of the pUC18 genes is for an enzyme
called β-lactamase. Possessing this gene
causes bacteria to be resistant to the antibi-
page 68
otic ampicillin. Bacteria lacking this plasmid will not grow in the presence of this
antibiotic. Acquiring this plasmid makes
the bacteria able to produce ß-lactamase and
able to grow in media that contains ampicillin.
Procedure:
1. We will be transferring bacteria into
sterile culture media, in tubes and on
agar plates. You want to avoid contaminating these media with other bacteria. The instructor will wipe down the
bench tops with disinfectant. Wash
your hands well before beginning the
procedure. The instructor will demonstrate how to transfer the bacteria without contaminating the culture ("sterile
technique").
2. Work in groups of 4. Obtain a sterile
microcentrifuge tube (hereafter called a
“tube”) to hold the bacterial culture
while we "soften it up" a bit. Your instructor may allow you to use the
micropipettes, or may designate certain
students to use them. Using sterile tips,
transfer 50µl of E. coli culture into each
tube, and then add 500µl of CaCl2 to
each tube.
3. Cap the tube and tap the end with the
tip of your index finger to mix the solutions. Then put the tube on ice for
about 20 minutes. This treatment improves the competence of the cells
(remember what that means?).
4. While you are waiting, obtain 2 more
sterile tubes and mark them "+" and "-",
for "plus plasmid" and “minus plasmid”. The instructor will add 10µl of
solution containing the pUC18 plasmid
to the "plus" tube.
5. Transfer 100µl of the competent cells to
each of the +/- tubes.
DNA Isolation and Genetic Transformation
6. Incubate the +/- tubes on ice for 30 minutes.
7. Transfer the +/- tubes to a 37C water
bath for 5 minutes. This heat shock facilitates the uptake of plasmid DNA.
8. Add 750µl of nutrient broth to each of
the +/- tubes and incubate at 37C for 30
minutes. This incubation period allows
the bacteria time to recover from the
CaCl2 treatment and to begin to express
the ampicillin-resistance gene on the
plasmid.
9. Follow your instructor's directions to
inoculate 4 agar plates. You should inoculate 2 plates from each of the 2
tubes- one plate with ampicillin and one
without. You will use a micropipette to
transfer 250 µl of the mixed bacterial
suspension from the tube to the plate,
and then use an inoculating loop to
spread the bacteria evenly onto the agar
surface. It is important to have areas
where the culture drop is is spread very
thinly, so that you can observe colonies
that grow from individual cells.
10. Leave the plates at room temperature
until the liquid has been absorbed into
the agar (about 10 minutes).
11. Tape the plates shut, then invert them
and write your name, lab section the
date, and the treatment on each. Incubate the inverted plates at 37C for about
24 hours.
12. After about 24 hours, you will have to
check your plates and count the bacterial colonies that have developed. Your
instructor will describe how to carry out
this procedure. Count the number of
visible colonies on each of your plates
and record these values in Table 1.
page 69
Table 1. Transformation Results
Treatment
Number of colonies
+ amp, - plasmid
+ amp, +plasmid
- amp, - plasmid
- amp, + plasmid
Assignment: Lab Report
Your report should focus on the transformation procedure. Follow the lab report format- it wouldn’t hurt to reread the description that appeared earlier.
In the introduction of your report, briefly
describe the bacterial genome and plasmids,
the mechanism of transformation, and the
overall rationale of the experiment. End the
Introduction with a statement of the predicted outcome.
State the Methods in your own words- don’t
transcribe the lab manual- and include any
important steps added by your instructor.
The Results section should have a written
description of the outcome as well as a table
giving the results.
In the Discussion, interpret the results, and
also extend your discussion to include information about either antibiotic resistance
plasmids or recombinant plasmids. You can
find more information in your text and the
sources that are linked to the Bio 121 lab
website. You will need to do some searching to find information about antibiotic resistance plasmids or recombinant plasmids.
Finally, don’t forget the Literature Cited
section.