Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Promoter (genetics) wikipedia , lookup
DNA barcoding wikipedia , lookup
Transcriptional regulation wikipedia , lookup
Comparative genomic hybridization wikipedia , lookup
List of types of proteins wikipedia , lookup
Silencer (genetics) wikipedia , lookup
Gene expression wikipedia , lookup
DNA sequencing wikipedia , lookup
Agarose gel electrophoresis wikipedia , lookup
Maurice Wilkins wikipedia , lookup
Molecular evolution wikipedia , lookup
Non-coding DNA wikipedia , lookup
Gel electrophoresis of nucleic acids wikipedia , lookup
Transformation (genetics) wikipedia , lookup
Vectors in gene therapy wikipedia , lookup
Biosynthesis wikipedia , lookup
Expression vector wikipedia , lookup
SNP genotyping wikipedia , lookup
DNA supercoil wikipedia , lookup
Nucleic acid analogue wikipedia , lookup
DNA vaccination wikipedia , lookup
Bisulfite sequencing wikipedia , lookup
Cre-Lox recombination wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Community fingerprinting wikipedia , lookup
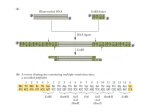
C l o n i n g a n d C o m p e t e n t C e l l s Design Genes with Ease Using In-Fusion® Cloning The work described in this article was performed at Harvard Medical School1 by B. Zhu, G. Cai, E.O. Hall, and G.J. Freeman and originally published in BioTechniques (1). 1 Dana-Farber Cancer Institute, Department of Medicine, Harvard Medical School, Boston, MA 02115, USA This article has been adapted from the original publication. In-Fusion can join any two pieces of DNA that contain a 15 bp overlap at their ends. The result is equivalent to a recombination event across the 15 bp overlap. The 15 bp overlap can be engineered by inclusion in forward and reverse primers used to PCR-amplify two segments of DNA. Originally described for inserting one piece of DNA into a restriction enzyme-digested plasmid, we have found that In-Fusion can efficiently join up to four pieces of DNA in a single reaction. We used this approach to construct seamless fusion proteins and modular vectors with readily interchangeable segments. In-Fusion-Ready DNA modules can be pieced together simultaneously without restriction digestion, regardless of sequence and size, thereby opening up new cloning possibilities. Visit our website DNA constructs are typically created by ligating two different DNA fragments that have been digested with restriction enzymes containing complementary restriction sites. Cloning options are often limited by the lack of available unique sites in the vector and gene of interest. In contrast, In-Fusion is an enabling technology that can join any two pieces of DNA that contain 15 bp of shared identity at their ends, without the need for any restriction enzyme digestion. The 15 bp overlap can be included in the primers used to PCR-amplify a segment of DNA. Moreover, the pieces of DNA can be generated either by PCR or by restriction digestion of plasmid DNA, regardless of the types of ends (sticky or blunt) produced. The In-Fusion mechanism is ligase-independent and uses the unique properties of poxvirus DNA polymerase (2–4). When incubated with linear duplex DNAs with identical ends in the presence of Mg2+ and low concentrations of dNTP, the 3'–5' proofreading activity of poxvirus DNA polymerase progressively removes nucleotides from the 3' end. This exposes complementary regions on the DNA fragments that can then spontaneously anneal through base pairing. This results in joined molecules containing a hybrid region flanked by nicks, 1–5 nucleotide gaps, or short overhangs. The annealed structures are moderately long-lived because the poxvirus DNA polymerase has a lower affinity for nicked or gapped DNA ends than for duplex ends. Introduction of this annealed DNA into Escherichia coli repairs any single-stranded gaps. In this study, we explore the unique end-joining capabilities of In-Fusion to combine not only two, but four fragments of DNA in a single, one-step reaction to develop seamless fusion proteins and modular expression vectors. Clontech Laboratories, Inc. A Takara Bio Company United States/Canada: +1.800.662.2566 • Asia Pacific: +1.650.919.7300 • Europe: +33.(0)1.3904.6880 • Japan: +81.(0)77.543.724 For Research Use Only. Not for use in diagnostic or therapeutic procedures. Not for resale. Clontech, the Clontech logo, EcoDry and In-Fusion are trademarks of Clontech Laboratories, Inc. All other marks are the property of their respective owners. Certain trademarks may not be r egistered in all jurisdictions. ©2013 Clontech Laboratories, Inc. 4.13 IN (630836) www.clontech.com C l o n i n g a n d C o m p e t e n t C e l l s Construct Design DNA sequence segments encoding the extracellular domain of murine (m) PD-L2 (5), the crystallizable fragment (Fc) of murine immunoglobulin G2a (IgG2a) (5), and the murine IgA tailpiece (6, 7), were assembled in silico with a mammalian expression vector using the Sequencher program (Gene Codes). The goal was to create a murine dodecameric immunoglobulin fusion protein (6, 7). Sense and antisense PCR primers were designed containing 15 bp of sequence which overlapped with the adjacent segment of the construct, as well as 20–30 bp of segment-specific sequence (see Figure 1 and Table I). The multiple pieces of DNA were joined seamlessly because the In-Fusion Enzyme does not add any additional nucleotides during the cloning reaction. All DNA fragments shown here were PCR-amplified from appropriate templates using overlapping primers that were designed as described in the In-Fusion Kit User Manual. Clontech® also provides an online tool for primer design. Fragments generated by PCR were amplified by a high-fidelity, hot start polymerase, gel-purified, and quantified by absorbance. The vectors used as backbones in this study were linearized using NcoI and SalI restriction enzyme sites. However, vector backbones with the desired 15 bp overhangs can also be generated by PCR using a highfidelity DNA polymerase (data not shown). In-Fusion reactions were set up according to the Clontech In-Fusion Kit User Manual. Between 25–100 ng of vector was mixed at a ratio of 1 mole vector to 2 moles of each additional DNA segment in 10 µl of water and added to one well containing the lyophilized In-Fusion reaction mix. After incubation, the In-Fusion reaction was diluted with TE and transformed into chemically competent E. coli according to the In-Fusion User Manual, miniprepped, and characterized by restriction enzyme digestion and sequencing. PD-L2 extracellular domain Table I: Primers for PCR Amplification of the PD-L2 Extracellular Domain, IgG2a Fc, and IgA Tailpiece PD-L2 Extracellular Domain with 15 bp Overlaps to NcoI-Digested Vector and IgG2a Fc, 693 bp PCR Product Pr o m NcoI er ot mlgG2a Fc Sense (NcoI site is underlined) 5'-TTCAAATCCACCATGGAGACAGACACACTCCTGCTAT-3' mlgA tailpiece Antisense 5'-CTTGATTGTGGGCCCTCTGGGGACTTTGGGTTCCATCCGA-3' IgG2a Fc, 678 bp PCR Product ) (A SalI Po ly Expression Vector Sense 5'-GGGCCCACAATCAAGCCCTGTCCTC-3' Antisense 5'-CCGGGAGAAGCTCTTAGTC-3' IgA Tailpiece with 15 bp Overlaps to IgG2a Fc and SalI-Digested Vector, 100 bp PCR Product Sense 5'-AAGAGCTTCTCCCGGCTGTCGGGTAAACCCACCAATGTCAG-3' Figure 1. Seamless construction of a three-domain immunoglobulin fusion protein with a four-piece In-Fusion reaction. Segments were generated by PCR using primers that contained a 15 bp overlap with the adjacent segment, and 20–30 bp of segmentspecific sequence. Colored regions indicate overlap regions with 15 bp of identity. Arrows indicate PCR primers. The segments were joined to a NcoI-SalI-digested expression vector in a ligasefree In-Fusion reaction. Antisense (SalI site is underlined) 5'-AGTAACGTTAGTCGACTCAGTAGCAGATGCCATCTC-3' Overlaps are colored to match Figure 1. 2 C l o n i n g a n d C o m p e t e n t C e l l s Multiple Fragment Cloning Results A major limitation of standard cloning approaches is the addition of unwanted amino acids that are encoded by the restriction enzyme sites used to join the DNA ends. This is particularly detrimental for fusion proteins and recombinant antibodies, since the undesired amino acids may perturb structure, reduce expression, or be antigenic. We generated a seamless murine PD-L2-IgG2a Fc-IgA tailpiece fusion protein by joining four pieces of DNA encoding the PD-L2 extracellular domain, IgG2a Fc domain, IgA tailpiece, and an expression vector containing a DHFR selection cassette. NcoI and SalI sites were used to linearize the vector and were therefore incorporated into the 5' and 3' segment fragments as part of the 15 bp complementary overhang with the linearized backbone (see Table I). No restriction sites were incorporated between the fusion protein domains. Since no restriction digestion of inserts is needed when using In-Fusion technology, the presence of NcoI and SalI restriction sites within the amplified fragments does not hinder cloning. Following the In-Fusion reaction, miniprep DNA was isolated from four colonies and analyzed by NcoI + SalI restriction enzyme digestion. All four minipreps were verified to contain all four pieces of DNA (data not shown). Two clones were subsequently sequenced and were found to be error-free isolates of the 1.5 kb coding region containing the PD-L2-IgG2a Fc-IgA tailpiece fusion (Table II). We then linearized the plasmid by MluI digestion, transfected the construct into DHFR-deficient CHO cells, and selected for DHFR expression. A production run yielded 12 mg/liter of purified 65 kDa fusion protein (Figure 2). This data shows that the In-Fusion technology can be used to seamlessly join three or more pieces of DNA to create a multimeric, highly expressed, soluble recombinant antibody. Discussion We observed fewer background colonies when the insertion site in a plasmid vector was created by digestion with two restriction enzymes rather than one. Treating the vector with phosphatase was unnecessary and reduced cloning efficiency. Accurate quantification and ratios of vector and DNA segments were the most critical factors for successful multipiece In-Fusion cloning. Of the17 constructs generated, 28 out of 38 sequenced clones were found to be errorfree, and none required sequencing of more than 3 isolates to obtain the desired sequence. The most common error found in incorrect isolates was a 1 or 2 bp deletion in a junction region, which could have resulted from an error in the repair reaction due to incomplete conversion of the 15 bp overlap region to a single-stranded form. The use of highly competent E. coli (1 × 109 cfu/µg) is recommended for multipiece In-Fusion reactions because the number of transformants decreases as the number of pieces of DNA in the In-Fusion reaction increases. Two-, three-, and fourpiece In-Fusion reactions resulted in an average of 671, 539, and 56 colonies, respectively, on a plate spread with 0.1 ml of a total 0.3 ml transformation reaction. In our experience, efficiency of the In-Fusion reaction does not depend on the length of the DNA segments, which have varied from 83 bp to 12 kb with little difference in efficiency. kDa 188– 98– 62– 49– 38– 1 2 Figure 2. Recombinant Antibody Protein Production. One µg of purified protein was analyzed using SDS-PAGE under reducing conditions and stained with Coomassie blue. Lane 1: Protein molecular weight standards with size in kDa indicated. Lane 2: Murine PD-L2IgG2a Fc-IgA tailpiece fusion protein. Table II: Transformation Efficiency of a Four-Piece In-Fusion Reaction PD-L2 Extracellular Domain IgG2a Fc IgA Tailpiece Vector 693 bp, 6.1 ng 678 bp, 6.0 ng 100 bp, 0.9 ng 11,365 bp, 50 ng Total Transformants No. ErrorFree/No. Sequenced 120 2/2 28– 17– 14– 3 C l o n i n g a n d C o m p e t e n t C e l l s Potential Applications Multifragment cloning via In-Fusion facilitates the construction of modular vectors (Figure 3) as well as mutations and synthetic genes (1). This can be accomplished by developing a toolbox of In-Fusion-Ready DNA modules that can be readily joined in infinite combinations. Fragments can include fusion protein partners [Fc, green fluorescent protein (AcGFP1), etc.], epitope tags, linkers, promoters, poly(A) sites, selectable markers, and origins of replication. Adjacent modules can be designed to contain complementary 15 bp overhangs to allow restriction enzyme-independent In-Fusion cloning of up to four modules simultaneously. If desired, unique restriction sites can be included in the primers used to amplify the modules. Additional features can be added to the modular vectors by incorporating restriction sites, translation initiation sites, linkers, or epitope tags in the primer sequences. Up to four pieces at a time may be joined by In-Fusion reaction, creating a modular vector whose individual components may be readily excised with restriction enzymes and/or replaced via In-Fusion. This study shows that In-Fusion can be successfully used for multifragment cloning. The fact that multiple amplified PCR products do not need to undergo any restriction digestion and can be used simultaneously for cloning into a linearized backbone by a one-step reaction opens up new cloning possibilities. Two-piece In-Fusion reactions have already been successfully used in high-throughput applications (4, 8). Adaptation of In-Fusion multifragment cloning for high-throughput applications is the next step to further unleashing the potential of In-Fusion technology. NcoI Promoter 1 and splice cDNA insertion site SalI Poly(A)-1 Modular Vector RS1 RS4 Figure 3. Modular vector. Red bars indicate junctions between segments. Different unique restriction sites (RS) can be engineered into the junctions via the primers used to amplify the segment. Each segment is amplified with primers that include 15 bp of overlap with the adjacent segment. DNAs are joined via an In-Fusion reaction, so no restriction digest is required. NcoI is a useful site because it includes a translation initiation site. MluI is a convenient site for linearization of the vector prior to transfection. Promoter 2 and splice RS5 ori, AmpR RS6 MluI Selectable marker Poly(A)-2 References 1. Zhu, B. et al. (2007) BioTechniques 43(3):354–359. 2. Evans, D.H. et al. (2003) In patent application 20030162265, USA, pp. 1–52. 3. Hamilton, M.D. et al. (2007) Nucleic Acids Res. 35(1):143–151. 4. Marsischky, G. & LaBaer, J. (2004) Genome Res. 14(10B):2020–2028. 5. 6. 7. 8. Latchman, Y. et al. (2001) Nature Immunol. 2(3):261–268. Hirano, N. et al. (2006) Blood. 107(4):1528–1536. Arthos, J. et al. (2002) J. Biol. Chem. 277(13):11456–11464. Berrow, N.S. et al. (2007) Nucleic Acids Res. 35(6):e45. Cat. # Product Package Size 638909 638910 638920 638911 In-Fusion HD Cloning Plus 10 Rxns 50 Rxns 96 Rxns 100 Rxns 638912 638913 638914 638915 In-Fusion HD EcoDry™ Cloning Plus 8 Rxns 24 Rxns 48 Rxns 96 Rxns Notice to Purchaser Your use of these products and technologies is subject to compliance with any applicable licensing requirements described on the product’s web page at http://www.clontech.com. It is your responsibility to review, understand and adhere to any restrictions imposed by such statements. Clontech products are to be used for research purposes only. They may not be used for any other purpose, including, but not limited to, use in drugs, in vitro diagnostic purposes, therapeutics, or in humans. Clontech products may not be transferred to third parties, resold, modified for resale, or used to manufacture commercial products or to provide a service to third parties without prior written approval of Clontech Laboratories, Inc. 4