Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Synthesis and Structure of Biomimetic Porphyrins
Brian Morgan and David Dolphin
Department of Chemistry, University of British Columbia, Vancouver, B.C., Canada V6T 1Y6
1.
Introduction ................................................................................................................................
2.
116
Porphyrins with Appended Peptides ...........................................................................................
121
3.
3. A.
3.B.
3.C.
3. D.
CheIatedHemes .........................................................................................................................
Porphyrins HavingCovalently Attached Imidazole or Pyridine Ligands .........................................
Porphyrins Having Covalently Attached Sulfur Ligands................................................................
Porphyrins with Covalently Attached Ouinonc Groups .................................................................
Porphvrins with Covalentlv Attached Interactive Groups ..............................................................
127
127
137
142
146
4.
4. A.
4. B.
"Picket-Fence" Porphyrins and Related Species ........................................................................
"Picket-Fence" Porphyrins............................................................................................................
"Tailed Picket-Fence" Porphyrins ................................................................................................
148
148
156
5.
5. A.
5.B.
5. C
"Capped" Porphyrins and Related Species..................................................................................
"Capped" Porohvrins .....................................................................................................................
"Pocket" and "Tailed Pocket" Porphyrins ......................................................................................
"Bis-Pocket" Porphyrins ................................................................................................................
160
160
166
167
6.
6.A.
6.B.
6.C.
6. D.
Strapped Porphyrins....................................................................................................................
Non-Functionalized Alkyl Straps....................................................................................................
Straps Containing Bulky Blocking Groups .....................................................................................
Straps Containing Interactive Groups............................................................................................
Doubly-StrappedPorphyrins ..........................................................................................................
169
169
175
184
192
7.
List of Abbreviations....................................................................................................................
198
8.
References .................................................................................................................................
199
1. Introduction
Because
have
ing
of
an
ubiquitousness
investigated
iron
storage
idases
on
porphyrin
(hemoglobin
reduction
in
heir
been
as
3
catalases) ',
and
contains
porphyrin
ring
occupying
their
diversity
of
an
variety
group,
four
hydrogen
porphyrin
must
protoporphyrin
coordination
by
the
sites
number
2
oxygen
destruction
(perox-
The
the
of
active
nitrogens
the
and
and
c) ,
P450)5.
IX),
contain-
transport
b,
and
proteins
all
oxygen
(cytochromes
utilization
heme
proteins,
for
(cytochrome
(usually
dictated
functions,
These
responsible
peroxide
planar
be
natural
levels.
transport
oxidation
essentially
function
their
are
electron
hydrocarbon
iron
of
interdisciplinary
prosthetic
oxidase) ,
case
the
and
myoglobin)l
4
each
fore
the
and
(cytochrome
and
and
multi-
metal.
nature
of
site
of
the
There-
the
axial
ligands, the spin and oxidation state of the iron and the nature of he polypeptide chain.
A
basic
tenet
biomolecules
sites.
iron
may
Obviously
porphyrins
oxidation
of
bioinorganic
be
to
mimicked
fully
understand
must
be
and
coordination
state
chemistry
using
the
undertaken
is
that
simpler
mechanisms
in
which
number)
the
inorganic
of
the
and
structure
and
complexes
to
heme
protein
characteristics
the
steric
of
and
function
model
function,
the
a
metal
electronic
of
large
the
active
study
(spin
effects
of
state,
of
the
porphyrin and other ligands are systematically varied.
Historically,
ism
of
much
reversible
five
of
the
research
oxygen
binding
coordinate
high
proteins
are
become
six-coordinate
low
spin
on
to
metalloporphyrins
myoglobin
and
spin
(S
=
iron(II)
=
0).
The
(S
2)
has
focussed
hemoglobin.
difficulty
species,
in
on
Oxygen
which
reproducing
the
mechan-
binding
upon
his
heme
oxygenation
behaviour
is
dominated by two problems:
(i)
(ii)
the irreversible oxidation of iron(II) porphyrins on exposure to oxygen, and
the difficulty in obtaining well-defined five-coordinate iron porphyrins.
Simple
iron(II)
porphyrins
cannot
reversibly
bind
oxygen,
except
at
low
temperature.
At room temperature, and in the absence of a large excess of a sixth ligand, formation of
the
six-coordinate
iron(II)
five-coordinate
iron(II)
breaks
presumably
down,
dioxygen
complex
via
to
a
species
is
immediately
give
the
μ-peroxo
ferryl
intermediate
to
give
followed
by
bisiron(III)
a
which the iron has been irreversibly oxidized to the ferric form (Scheme I)8-11.
u.-oxo
attack
of
complex.
bisiron(III)
a
second
This
rapidly
complex
in
117
Therefore,
the
a
oxygen
irreversible
porphyrins
must
major
binding
oxidation
is
role
site,
via
possible
provide
an
the
enzyme
to
incapable
of
oxygen
exists
the
body
pathway
formation
is
superoxide13'.
chromes
occurs
inhibited,
Similar
P45014
and
close
transport)
in
in
the
complex.
to
occur
the
form
acid
believed
cytochrome
is
to
with
oxidase3.
of
of
That
the
the
The
by
(the
about
of
chain
to
so,
of
hemo-
alternative
complex
protonated
for
stabilizes
body
hemoglobin
μ-peroxo
superoxide,
iron(II)
the
This
formation
also
of
that
Even
3%.
sheath
consequent
iron(III)
where
assisted
production
fact
form12.
of
is
and
oxidation
oxidized
conditions
proton
peptide
rings
the
iron(II)
extent
under
involve
proteins
heme
irreversible
functional
to
or
heme
two
demonstrated
methemoglobin
ferric
aqueous
is
backbone
approach
mechanism
reduce
oxidations
and
polypeptide
the
[μ-peroxo
ano her
being
oxidation
the
by
globin
in
for
preventing
the
the
cytoFe11-O2
species by
enclosing
the
porphyrin
in
a
hydrophobic
pocket
to
which
access
In addition, recent neutron15 and X-ray diffractionl6 studies have indicated that stabili-
by
protons
is
inhibited.
118
zation
to
of
the
hydrogen
iron-oxygen
bonding
bond
between
in
oxyhemoglobin
the
terminal
and
oxygen
oxymyoglobin
atom
and
may
the
in
part
imidazole
be
of
due
the
distal
histidine (His-E7).
The
influence
barrier
to
believed
to
Similarly,
provide
the
of
the
oxidation.
be
the
an
protein
Conformational
responsible
arrangement
avenue
protein's
of
along
role
in
tor
backbone
is
changes
upon
the
residues
electron
maintaining
the
than
binding
cooperativity
on
transfer
pervasive
oxygen
remarkable
certain
which
more
the
may
coordination
simply
at
the
exhibited
protein
occur
in
sphere
of
has
the
providing
active
been
iron
are
hemoglobin17.
by
postulated
cytochromes.
the
a
site
But
to
it
porphyrin
is
which
determines the functions of the various heme proteins.
The
metal
second
for
N-donor
ria
in
major
problem
six-coordination.
ligands,
Eq.
9,
in
studying
For
six-coordination
>
K2
K1
(K2/K1
simple
example,
is
=
iron
in
favoured
10-30 in
porphyrins
solution
over
is
the
containing
five-coordination,
aprotic solvent
25°C)18.
at
preference
strongly
of
the
coordinating
i.e.,
for
In
benzene
the
equilib-
at
0
25
C
the
binding
constants
of
4
and K2 ~1.9 x IO
the
iron.
Addition
second
field
The
of
pyridine
ligand
ligand
to
stabilization
have
been
estimated
at
K1
~
1.5
IO3
x
M-1
M '. The size of K1 and K2 is obviously controlled by the spin state of
four-coordinate
one
Fe11(TPP)
to
-
form
iron
gives
the
energy19-20.
the
porphyrin
high
low
spin
In
is
spin
(S
(S
=
contrast,
0)
for
in
an
=
2)
intermediate
six-coordinate
Co",
spin
five-coordinate
no
species
(S
=
1)
complex
which
with
gain
stabilization
a
is
gained
state.
adds
a
in
crystal
on
going
from five- to six-coordinate since Co" is low spin in both cases, and K1 > K221).
A
nate
fur her
iron
which
greater
the
iron
ligating
bis(imidazole)
difficult.
consequence
porphyrins.
is
A
this
power
of
by
an
imidazole
makes
which
is
example
coordinated
complexes
Strategies
of
typical
self
control
the
is
difficulty
the
imidazole
(His-18)
coupled
with
assembly
of
coordination
of
preparation
are
its
the
preparing
of
mixed-ligand
models
and
a
essential
to
ligand
for
six-coordi-
cytochrome
thioether
tendency
mixed
for
form
in
The
six-coordinate
system,
preparing
c
(Met-80).
a
Im-Fe-SR2,
range
of
heme protein model porphyrins.
Numerous
approaches
have
been
used
to
control
oxidation
and
coordination
in
model
oornhvrin svstems.
(i)
Excess
Ligand:
The
presence
of
excess
base
(imidazole,
concentration of five-coordinate heme and reduce u-peroxo complex formation.
pyridine)
will
minimize
the
119
(ii)
Temperatures22-'25.
Low
atures
one
(~-60°C),
is
Iron(II)-O2
where
reduced
to
the
porphyrin
irreversible
studying
competitive
complexes
oxidation
oxygen
are
reactions
binding
stable
are
K2/K1
as
at
low
slowed
temper-
down.
increases
as
Again
temperature
decreases.
(iii)
Kinetic
ible
oxygen
Ior
has
of
subjected
then
binding
even
exploited
solution
Fast
Measurements:
the
to
a
short
of
equilibrated
laser
preferentially
pulse
wi h
me hods
conditions
stability
Im-Hm-CO,
reacts
spectroscopic
under
where
may
imidazole-heme-CO
with
a
which
oxygen
mixture
dissociates
at
a
fast
be
irreversible
complexes
of
oxygen
the
but
used
to
oxida ion
carbon
observe
will
towards
and
A
monoxide,
is
The
(kBO2
rate
>
Tray-
oxidation26.
carbon
monoxide.
measurable
revers-
occur.
deoxy
IO7
heme
M-'s-1).
In
IO3 — 10s, the Im-Hm-O2 complex dissociates and returns to the Im-Hm-CO complex.
Since
kBCO
determined
is
in
the
the
rate
of
experiment
return
and
kB
before
2
O2
02
and
k^
is
may
added,
be
kB
CO
calculated
may
from
a
be
accurately
plot
of
l/krelurn
vs O2 (pressure).
(iv)
Metal
proper ies.
Co
and
Replacement
Substitution:
metalloporphyrins
which
Such
Ru
are
an
more
approach
porphyrins.
In
is
the
of
iron
inert
to
applicable
case
of
cobalt28
with
oxidation
since
Co,
and
apoproteins
reconstituted
ruthenium29
or
possess
Co
different
may
be
leads
to
coordination
recons ituted
hemoglobin
exhibits
with
coopera-
tive oxygen binding although to a diminished extent.
(v)
This
Immobilization:
anchoring
diethyl
The
the
ester
matrix
porphyrin
or
have
contained
a
the
Fe (TPP)(B)2
axial
base
attempts
ligand
may
the
a
be
latter
(pyridine
observe
tion of oxygen by the silica support.
Wang's
of
approach
uptake
covalently
attached
approach
attached
and
piperidine)
reversible
to
oxygen
and
two
to
in
the
binding
rigid
a
also
support.
silica
flowing
Basolo
gel
obscured
and
support
removed
iron(II)
by
the
a
either
Reaction
helium
five-coordinate
was
by
heme
provided
Alternatively,
surface31.
the
heating
give
a
a
l-(2-phenylethyl)imidazole.
but
observed.
prepared
oxidation
experiment30
and
hemes
was
to
irreversible
classic
polystyrene
of
oxygen
porphyrin
or
prevent
In
matrix
groups
the
to
support.
close
Reversible
undertaken
to
in
prevented
coordinated
attempts
solid
embedded
only
3-imidazolylpropyl
11
However,
approach
to
environment.
colleagues
sixth
was
not
hydrophobic
the
porphyrin
his
which
with
the
porphyrin.
physisorp-
120
(vi)
Steric
approach
vented.
rin
two
The
ring
in
prepare
approach
a
sterically
porphyrin
rings
most
polymer
compounds
temperature.
By
encumbrance:
of
closely
chain.
capable
However,
doubts
the
approach
has
reversible
about
one
or
therefore
mimicking
This
of
blocking
and
the
both
bridge
natural
system
been
oxygen
number
faces
u-oxo
and
is
vigorously
binding
the
to
porphyrin,
aqueous
of
the
may
enfold
pursued
in
nature
of
formation
in
the
an
pre-
porphy-
attempt
solution
active
close
be
at
sites
to
room
and
the
reversibility of oxygenation have made this approach less fruitful.
In
are
contrast,
obstructed
porphyrins
by
have
some
been
group(s)
synthesized
covalently
in
which
bonded
to
one
the
or
ring.
both
The
faces
function
of
of
the
ring
the
steric
bases
which
hindrance is two-fold:
(i)
to direct base binding to the open face, ensuring five-coordination, and
to allow O2 to bind on the hindered face, steric hindrance preventing u-oxo bridge
(ii)
formation.
Five-coordination
may
also
be
ensured
in
these
systems
by
using
bulky
axial
cannot bind on the protected face.
This
ally
approach
different
has
been
model
used
by
many
porphyrins,
groups
e.g.
to
produce
picket-fence,
a
wide
variety
capped,
of
architectur-
cyclophane,
crowned,
strapped, basket-handle, etc., which are discussed below.
(vii)
Chelated
allows
one
thioether,
of
(Eq.
Covalent
Hemes:
control
phenoxide,
likelihood
ligand
to
the
etc.,
coordination
15).
As
attachment
extent
of
covalent
to
the
long
as
of
attachment
metal
the
coordination.
increases
without
displacement
ligand
For
the
does
the
necessity
not
to
poor
local
of
occur,
the
porphyrin
ligands
such
periphery
as
thiolate,
concentration
a
large
addition
excess
of
a
and
of
the
external
second
ligand
allows formation of six-coordinate mixed ligand systems.
On
built-in
six-
he
other
1:1
and
hand,
for
base/porphyrin
strong
ligands
stoichiometry.
four-coordinate
(Eq.
16),
As
is
e.g.
imidazole,
long
as
prevented
pyridine,
chelation
dimerization,
this
to
approach
form
produces
provides
mixtures
a
of
five-coordinate
complexes.
In
the
following
brance
and
chelation
porphyrins
fixed
sections
as
we
models
hat
have
been
distances
from
the
for
examine
those
heme
proteins.
synthesized
porphyrin
in
ring.
order
transfer
between
various
components
gained.
Several
excellent
reviews
exist
these
models
and
their
congruency
wi h
the
also
create
these
of
which
We
to
Using
energy
porphyrins
the
discuss
natural
which
review
employ
a
series
complexes
models,
photosyn hetic
the
with
important
ligand
of
encum-
biomime ic
quinones
information
apparatus
binding
systems20,32-37.
concentrate here on the strategy and synthetic details of model porphyrin preparations.
steric
can
properties
Instead,
we
at
on
be
of
will
121
To
this
end,
the
compounds
have
been
grouped
toge her
more
in
terms
of
structure
than
of function.
The reader is directed to reference 56 for a discussion on porphyrin dimers and strati
bisporphyrins which are not reviewed here.
2. Porphyrins with Appended Peptides
Perhaps
duce
the
the
tide
most
local
fragments
acids
(e.g.
obvious
approach
environment
to
a
histidine,
of
to
the
suitable
the
porphyrin.
methionine),
synthesis
heme
active
If
the
reproduction
of
site
heme
by
peptide
of
the
protein
covalently
models
attaching
is
to
repro-
various
pep-
fragments
contain
suitable
amino
coordination
sphere
of
heme
the
protein may be possible.
An
early
example
histidine-containing
(Scheme
histidyl
2).
imidazole
via
sulfide
his
tripeptides
After
histidine-containing
of
metal
was
to
to
give
the
insertion
possible
pep ides
linkages
approach
were
was
propionic
into
depending
attached
2(b-d),
that
the
on
to
of
Lautsch
acid
the
al.38',
side-chains
porphyrin,
the
et
length
ethyl
a situation similar
of
intramolecular
of
side
to that
the
Miiller39' coupled L-histidine methylester and protohemin 3 with dicyclohexylcar-
coupled
coordination
peptide
chains
in
who
mesoporphyrin
of
chain.
various
IX
by
Similarly,
mesoporphyrin
cytochrome c.
la
the
Losse
IX
and
122
bodiimide
in
histidine
for
N,N-dimethylformamide.
bound
to
coordination
various
the
of
di-
the
time
Warme
imidazole
tripeptide
to
protohemin
chlorocarbonate
to
yield
groups.
The
mesohemin
sulfuric
anhydride,
been
converted
histidine
or
(Scheme
4).
methionine
paration
A
and
intramolecular
min
8
with
L-histidine
mixture
of
ester
bis(histidine
the
ester
three
9c)
methyl
to
was
ester)
9b,
and
was
tedious
and
have
more
The
obtained
in
yields
too
histidine
and
the
side
chains
had
acids
and
and
pre-
low.
also
It
a
and
as
a
(Scheme 5). The reduced iron(II) species was capable of binding oxygen reversibly at low
unstrained
five-coordinate
of
triethylamine,
deuterohemin
was
allow
Treatment
deuterohemin
as
7a-e
histidine
to
gave,
such
hemins
Unfortunately
syn hesized
yield
same
yielded
both
were
and
the
5
amino
short
triethylamine
16%
At
disubstituted
properties.
desired
unreacted
acid
prepared.
and
L-Ala-L-His;
3).
containing
short
coupled
triethylamine
complex
with
single
too
al.40',
appended
or
also
were
al.42),
of
propionic
reaction
a
was
et
(Scheme
mono-
ethylchloroformate
9a-c.
from
the
with
chain
Gly-L-His;
SO3/DMF
7e,
coordinating
of
Heijden
4a-d
was
arms
et
the
dihydrochloride
separated
ei her
quite
der
presence
a
of
model
side
Momenteau
compounds
both
porphyrin
was
side
containing
with
Subsequent
c
the
products
quantities
or
yielded
cytochrome
characterized
equimolar
porphyrins
one
peptide-containing
and
methyl
which
the
derivatives
6
that,
the
P-Ala-His;
in
mesohemin
anhydride.
coordination.
porphyrin
bispeptidyl
of
Van
(e.g.
DMF
ester
of
the
in
indicated
length
centre.
fragments
sulfonic
attached
the
iron
methyl
potential
isolation
that
iron
methyl
a
covalently
recognized
a
into
the
3
of
in
models
acid,
prepared
reaction
methionine
to
the
Hager41
and
methionine
However,
propionic
and
Gly-L-His-Gly-OEt)
ethyl
porphyrin
after
deuterohefollowed
by
purification,
6(7)-mono-(histidine
deuterohemin
mixture
of
6,7-
isomers
123
124
temperature
but
dimerization
oxidized
of
the
irreversibly
5-coordinate
at
room
species
temperature.
occurred
at
low
Furthermore,
temperature
extensive
(-60°C),
com-
plicating oxygen binding studies.
A
similar
dimerization
was
observed
for
the
iron(III)
species
at
room
systems
which
temperature
in
6-coordinate,
or
concentrated solutions.
To
his
mixtures
of
prepared
after
the
all
5-
porphyrin
deuterohemin
solvent
stage
for
8
or
the
and
syntheses
6-coordinate
having
mesohemin
la
purification.
The
Controlled
yielded
species,
derivatives
hours43).
three
have
two
with
bis-chelated
hydrolysis
with
model
separation
of
covalently
excess
M
10
are
could
be
attached
imidazoles,
in
in
histamine
product
2
which
was
hydrochloric
vacuo
obtained
acid
in
gave
tedious.
Castro
by
heating
the
absence
up
to
50%
a
20%
yield
of
yield
of
the monochelated hemin 11, again as a mixture of isomers (Scheme 6).
More
recently,
(obtained
prepare
and
from
a
hemin
triethylamine.
protohemin
series
was
The
Molokoedov
of
dibenzyl
al.44,
have
ester
12
histidine-containing
completed
yields
et
of
by
the
product
mixed
used
in
peptide
61%
from
yield
derivatives
anhydride
decreased
protohemin
method
47%
to
by
monobenzyl
partial
14a-e.
using
25%
Coupling
ethyl
as
peptide chain increased, the products being obtained as a mixture of the 6- and 7-isomers
ester
hydrolysis),
of
length
to
peptide
chloroformate
the
13,
of
and
the
125
(Scheme
7;
tide
heme
14e
only
the
6-isomers
derivatives
are
were
shown).
reported
to
The
reduced
be
stable
series
of
pentapeptide
for
35-40
14d
and
hexapep-
at
room
tempera-
minutes
ture in chloroform solution in the presence of air.
This
work
was
appended
peptide
derivatives
(a
ments,
bisaminoacyl
the
of
protohemin
histidine
more
the
prepare
6-
IX
8).
and
hydrophobic
previously
were
mixed
anhydride
derivatives
in
the
15a-i
as
spectra
peptide
environment
protohemin
prepared
7-isomers)
or
Absorption
residues
a
The
pentafluoroester
(Scheme
tuted
to
fragments45.
mixture
using
(30-60%)
extended
of
the
condensed
a
mixture
bulky
pep ide
the
of
chain
with
peptide
the
6-
frag-
unsymmetric
and
7-isomers
having
unsubsti-
pentacoordination.
was
two
monoaminoacyl
give
complexes
capable
15
IX
various
to
of
that
were
derivatives
with
methods,
indicated
chain
IX
protohemin
believed
Further-
to
enhance
the stability of the 5-coordinate iron(II) derivatives.
There
peptide
have
be
invariant,
the
polypeptide
imidazole
teau
been
of
structure
via
His-18
then
metal
by
are
covalently
synthetic
one
IX
strategy
(16)
optical
centre
mimic
thioethers
bonds
to
of
of
the
poor
the
of
to
that
the
that
a
sphere
for
two
ligands
the
obtain
Binding
ligands
to
indicated
coordina ion
attaching
porphyrin,
to
spectra
17.
the
active
c
The
methyl
observed
attempt
thioether
to
the
(TPP)
cytochrome
residues
for
iron
the
of
Lautsch
ethyl
side
in
porphyrin.
was
natural
to
In
this
to
and
17,
while
the
the
heme.
attached
were condensed with the TPP derivative 18 bearing a propenoate ester side chain at a (b-
9).
could
the
cytochrome
a
protected
2,4-a,a'-dib-
(Scheme
to
Momen-
a
of
overcome
Various
to
bound
histidine
case
heme-
14-18
is
methionine,
made
used.
17
using
site
chains
the
c
segment
of
al.38,
et
of
e.g.
the
was
14
atom
porphyrin
imidazole
the
active
cysteine
thioether
An
of
reveals
heme
appended
the
iron.
tetraphenylporphyrin
site
cytochromes
he
the
similar
(Cys-Gly-Gly-Cys-His)
insertion,
model
various
14-Cys-X-X-Cys-His-18.
sulfide
a
to
of
provides
with
romomesoporphyrin
the
attempts
Sequencing
with
Loock46,
and
pentapeptide
iron
two
compounds.
After
bind
iron
c.
this
cysteine
However,
poor
deriva ive
to
would
binding
of
the
dipeptides
126
127
pyrrole
position
residue
with
atropisomers,
on
the
the
1
Reaction
cis-endo
peptide
19
chain
porphyrin.
H-NMR
of
a
dipeptide
containing
cis-meso-tetraphenylporphyrin-3-propenoic
are
When
and
cysteine
residue
the
substituted
Zn
cis-exo
disposed
the
magnetic
the
and
circular
might
in
dipeptide
be
porphyrin,
20
(Scheme
a
favourable
chain
10).
In
the
suggested
However,
that
a
recent
interaction
mixture
cis-endo
case,
for
binding
(R
metal
EXAFS
could
S-alkvl
a
Gly-(SR)Cys-OEt
dichroism
Zn-sulfur
terminal
gave
conformation
was
occurring.
the
a
acid,
only
to
a
Me,
metal
bond
weak
in
trityl),
involving
indicated
be
two
substituents
=
sulfur
data
cysteine
of
that
and
for
long
range, if it occurred at all50.
3. Chelated Hemes
3. A. Porphyrins Having Covalently Attached Imidazole or
Pyridine Ligands
Chang
and
imidazole
bond51.
imidazole
would
Traylor
had
On
the
21
wi h
give
argued
either
a
too
other
the
acid
strain-free
that
few
hand
in
or
heme-pep ide
too
it
chloride
many
was
of
models
atoms
argued
pyrroporphyrin
five-coordinate-system
23
to
that
XV
the
side
chains
achieve
a
strain-free
condensation
22
(Scheme
followed
11).
was capable of binding dioxygen in a reversible manner in the solid state or when
of
by
This
containing
the
iron-imidazole
l-(3-aminopropyl)
insertion
"chelated"
of
iron
heme
128
dissolved
-450C,
in
a
only
tives
of
polystyrene
irreversible
pyrro-,
the
porphyrin
and
mesoheme,
and
1356.
purified
In
or
protohemin
3
base-containing
methanol.
pared.
The
of
25
was
having
amide
or
hemes
binding
pyridine
were
easily
dimethyl
then
coupled
to
using
with
30%
26,
yield.
28
The
24
were
as
was
amine
followed
was
then
isolated
dichelated
in
solution
series
of
covalently
shown
in
partially
or
Alternatively
chloride
A
investigated26,53-55.
prepared
primary
mixture
reversibly
imidazole
was
ester
chloride.
pivaloyl
reaction
products
to
a
pivaloyl
excess
The
or
linkages
porphyrin
oxygen
temperature52.
room
the
amine.
up
of
at
mesoheme
monochelated
in
capable
occurred
ester
base
treated
primary
isomers
While
monochelated
approach
pyridine
was
and
through
the
one
monoacid
imidazole
mixture
protoring
film.
oxidation
by
one
containing
were
an
of
the
water
or
chromatography
compounds
12
The
available
equivalent
with
to
proto-
Schemes
commercially
quenched
by
bound
For
hydrolyzed.
alcohol
at
deriva-
as
similarly
a
pre-
129
The
versatility
kinetics
side
of
the
chains,
of
O2
the
the
chelated
and
CO
chelated
heme
binding
base,
and
approach
to
has
these
the
leng h
allowed
compounds.
and
nature
the
systematic
Changes
of
the
in
study
solvent,
chelation
arm
of
the
porphyrin
have
been
correlated with changes in the association and dissociation rates of O2 and CO57-58.
Unlike
tion
the
"chelated-histidine"
at
low
temperatures,
(Scheme
14)
at
Ior
exploited
iron(lll)
he
such
dimerization
IX
binding
the
addition
of
titration
of
side-chain
the
titration
gave
a
straight
to
greatly
system
29,
the
side
of
(Scheme
30.
A
slope
n
=
While
2.1,
too
and
cyanide
Tray-
For
short
he
to
allow
binds
very
vice
versa.
Therefore
clean
conversion
leads
to
vs.
log
Y/l-Y
indicating
forms
Indeed,
cooperativity59.
pyridine
(log
dimeriza-
polymeric
studies26.
is
affinity
with
plot
any
his
chain
15).
29
in
underwent
of
exhibiting
pyridine
Hill
which
presence
used
a
protohemin
dimer
the
concentrations
increases
pyridine
line
against
design
pyridine
cyanide
pyridine-hemin-CN_
the
and
Momenteau42
of
argued
derivative
of
hemin,
to
Traylor
temperatures
protoporphyrin
intramolecular
system
poorly
[CN])
cooperativity
between
symmetric
diheme60.
for
to
this
the
metal
centres.
Axial
base
chelation
was
di-(3-pyridyl)ethylenediamine
ester
32
through
(Scheme
16).
the
The
similarly
(31)
pivaloyl
used
was
anhydride,
reaction
with
to
prepare
coupled
CO
followed
by
exhibits
two
a
with
iron
insertion
rate
Meso-1,2-
mesoporphyrin
to
give
constants,
monomethyl
the
diheme
indicating
either
33
two
environments or a sequential change of environment due to cooperativity.
The
protohemins
orientation
shifts
1
paramagnetic
for
published
34
of
and
the
H-NMR
35
base
the
methyl
for
various
spectra
with
and
of
studied61.
were
respect
vinyl
heme
to
protons.
proteins
the
imidazole-cyanide
Chelation
the
of
the
porphyrin
Comparison
provided
ring,
of
complexes
imidazole
causing
these
for
dichelated
a
different
shifts
confirmation
of
maintains
with
the
fixed
chemical
the
values
heme-imidazole
orientations proposed in the natural systems.
Tabushi
heme
et
groups
densation
(22%)
of
(Scheme
al.,
in
have
gable
the
chelated
62-64
c3
.
The
heme
approach
"gable-porphyrin"
(m-formylphenyl)triphenylporphyrin
17).
metal
insertion,
38
and
g,g'_pyridylmethane
porphyrins
the binding of I-MeIm and CO.
wi h
bridging
the
36
After
N,N'-diimidazolylmethane
stable
used
cytochrome
ligands
use
which
with
of
39
to
37
mimic
was
the
pyrrole
dimeric
resulted
displayed
orientation
prepared
and
bridging
in
by
of
the
the
con-
benzaldehyde
ligands
such
as
the
formation
of
cooperative
behaviour
to
130
131
1
The
dimers
H-NMR
or
chlorophyll-a
mate.
analysis
higher
and
converted
Reaction
chlorophyll
"chelated
with
derivative
magnesium
of
chlorophyll
aggregates.
chlorophyll"
mixed
he
complicated
Sanders65
to
(Scheme
18).
aggregation
42
often
and
was
and
used
by
with
40
Intramolecular
by
its
tendency
to
he
phytyl
group
removed
anhydride
l-(3-hydroxypropyl)imidazole
41
prevented
it
is
Denniss
triethylamine/ethyl
resulted
binding
gave
well
resolved
Boxer
and
Wright
in
of
1
the
to
he
spectra.
model
the
of
chloroforchelated
imidazole
H-NMR
form
to
the
A
similar
complex
formed
when apomyoglobin is reconstituted with chlorophyll derivatives66.
Momenteau
et
al.
this
series
the
base
ring,
via
amide
or
have
is
synthesized
attached
ester
to
linkages
afforded
the
monoformyl
derivative
to
the
acrylate
as
tion
yield
and
saponification
sponding
pyridine
45
acid
49
gave
chloride
resulted
in
47
he
a
a
44,
cis
chelated
of
Vilsmeier
was
and
acid
of
position
19).
which
of
propionic
with
series
(3-pyrrole
(Scheme
mixture
the
similar
the
chelated
elaborated
by
trans
isomers.
derivative
46.
porphyrin
heme
a
of
Cu(TPP)
Wittig
Demetallation,
Treatment
48
50
compounds67.
tetraphenylporphyrin
formylation
l-(3-aminopropyl)imidazole
appropriate
a
or
of
In
(TPP)
(43)68
condensation
hydrogenathe
corre-
or
3-(3-hydroxypropyl)
51
(70%
respectively) (Scheme 19). These models were used to study the kinetics of base binding
and
60%
132
to
4-
and
5-coordinated
TPP69),
iron(II)
and
also
to
study
the
by
Eaton
et
transient
oxygenation
of
iron(II) carbonmonoxy TPP after photolytic displacement of CO70.
The
acid
chloride
labelled
was
same
of
TPP-acrylic
acid
Cu-TPP
acrylic
Cu-TPP
investigated
derivatives
using
EPR
system
acid
52b-f
by
was
52a
wi h
(Scheme
varying
used
the
a
20).
nature
nitroxyl
The
of
al.71,72
resulted
extent
the
of
nitroxyl,
in
the
Treatment
metal-nitroxyl
the
of
appropriate
linkage
the
spin-
interaction
(amide
or
ester), and the geometry of the complex (cis or trans). A similar study was carried out on
a vanadyl porphyrin73,
133
Collman
to
the
al.74),
et
benzaldehyde
(54)
chromatography,
with
have
position
ortho
and
of
stannous
chloride
various
imidazole
pounds
58a-c
as
chains
prepared
of
a
pyrrole
chelated
meso-phenyl
(55)
the
in
compounds
where
Condensation
of
glacial
acetic
acid
the
chain
he
(Scheme
21).
for
the
mono-o-amino-TPP
Collman
less
and
readily
his
gave
(57)
which
colleages
accessible
a
attached
2%
was
merely
(53),
yield
56.
"tailed-picket
rins which are discussed in Sect. 4B. Mashiko et al.75 used the same chelated TPP
is
o-nitrobenzaldehyde
meso-mono-(o-nitrophenyl)triphenylporphyrin
produced
substitutes
hot
TPP
ring.
after
Reduction
coupled
used
the
fence"
with
com-
porphy-
134
compounds
els
for
to
frustrated
by
covalently
ligand
tem.
control
cytochrome
the
coordination
c,
which
in
greater
affinity
the
imidazole
attaching
a
contain
was
provided;
addition
These
authors
prepared
of
of
mixed
histidine
heme
to
the
thioether
several
ligand
and
iron
for
porphyrin
then
complexes
system.
methionine
imidazole
ring
furnished
with
Attempts
as
a
the
different
to
the
rather
axial
than
stoichiometric
mixed
tail
prepare
mod-
ligands,
are
thioether.
By
amount
six-coordinate
lengths
and
of
sys-
various
thioethers. For the Cs tail with tetrahydrothiophene as the thioether, a crystalline
iron(II) complex
structure
determined
(59) (Scheme 22) was obtained and its crystal
(Fig.
1).
Efforts
to
obtain
the
corresponding
were defeated by "head-to-tail" dimerization.
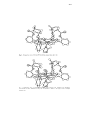
Fig. 1. Computer produced perspective of Fe11(C5Im)(TPP)(THT) 59. Adapted from Ref. 75
iron(III)
complex
135
A similar chelated TPP compound 60 was used by Walker to study the effect of axial
ligand
plane
orientation
of
axial
complexes .
ligand
In
bond
a
pyridine
ligand
bound
(57)
to
H-NMR
this
strain.
(o-aminophenyl)triphenylporphyrin
ing
1
the
76
(TPP)bis(imidazole)
effect
on
a
In
shifts
case
of
the
addition,
to
prepare
zinc
TPP77).
pyrrole
was
Walker
a
1
the
tail
and
series
H-NMR
made
and
protons
shorter
Benson
of
iron(III)
study
have
derivatives
visible
in
to
used
61a-e
spectroscopy
the
monocontain-
were
used
to study the displacement of the 3-pyridyl ligand by free 3-picoline (Scheme 23).
Condensation
porphyrin
6378).
common
N-I
deprotonation
of
In
this
position
addition
57
with
case
to
of
the
allow
trans-urocanic
imidazole
deprotonation
Cu(acac)2
acid
was
yielded
to
chloride
attached
the
the
at
(62)
the
imidazolate.
furnished
C-4
rather
After
[μ-imidazolato
(Scheme 24), a poten ial model for he [Cuu2+/Cyta33+] center of cytochrome oxidase.
iron
binuclear
the
than
chelated
the
more
insertion
and
complex
64
136
However
magnetic
essentially
and
EPR
studies
non-interacting,
unlike
al.79
used
of
the
64
indicated
strongly
that
FeII
the
2+
coupled
CuII
and
3+
[Cuu /Cyta3 ]
pair
centers
in
the
were
natural
system.
Molinaro
attach
a
et
series
of
pyridine
0-hydroxyphenyl-TTP
chelated
(68)
the
(Scheme
(-
500C
Goff
The
to
80°C)
Reaction
of
-
used
K2CO3
the
of
or
length
or
40%
but
72,
the
ester
the
which,
elongation
of
the
of
to
(65)
with
allowed
axial
a
to
react
TTP
may
iron-imidazole
was
prepare
chelated
"tension"
via
be
bond.
an
(65)
ether
of
65
elaborated
with
base
did
not
dibromoalkane
derivative
73
into
This
was
the splitting and shift of the pyrrole resonances in the 1H-NMR spectrum.
in
27%
overall
at
low
temperatures
oxygen
to
have
affinity.
the
corre-
solvent
26).
molecule
yield
imidazoles80.
forms
DMF
(Scheme
the
found
of
the
in
appended
(71)
imidazole
introduced
furnished
enhance
with
covalently
Reaction
methyl-4-bromobutyrate
70
oxygen
porphyrins
with
(66)
and
to
to
linkage.
hydrobromide
reversibly
the
strategy
required
chain,
ring
condensation
which
reacted
presence
when
the
alkane
porphyrin
The
(69),
derivatives
synthetic
gives
the
yield.
butoxy
cobalt
to
3-(bromoalkyl)pyridine
o-hydroxyphenyl-TTP
Et3N),
of
with
in
same
ether
tilting
67
porphyrin
25).
sponding
the
(65)
porphyrin
gave
mono-(o-hydroxyphenyl)tritolylporphyrin
ligands
By
in
an
(using
varying
the
effect
form
on
137
3.B. Porphyrins Having Covalently Attached Sulfur Ligands
Because
of
cytochrome
large
heme
the
poor
affinity
P450
have
usually
consisted
of
excess
mercap ide
concentrations
approach
available
for
to
binding
covalently
to
the
of
attach
metal
iron(II)
porphyrins
of
ion.
solutions
However,
mercaptide
without
for
of
the
to
the
mercaptide
porphyrins
Traylor81
has
porphyrin
necessity
of
anion,
models
of
the
presence
of
in
used
periphery,
excess
(Scheme 27). Protohemin chloride monodimethylamide monoacid (74) was coupled to 1-
the
chelated
making
external
it
ligand
138
amino-3-mercaptopropane
addition
CO
of
anion
in
formation
resulted
pound
he
79,
benzoyl
dimsyl
containing
analogous
CO
ester
(77)
and
of
the
two
masked
complex
80.
(75).
After
warming
reduction
removed
the
carbonmonoxy-mercaptide
mercaptides
Protection
also
the
mercaptide
sodium
group,
complex
was
of
with
benzoyl
prepared
as
dithionite,
and
78.
addition
A
similar
and
deprotected
the
benzoylthio
of
com-
to
give
derivative
before reduction of FeIII was necessary because of the reducing ability of mercaptans.
Alternatively
resultant
protohemin
82
disulfide
was
was
coupled
treated
with
with
sodium
bis(3-aminopropyl)-disulfide
dithionite,
the
iron
being
(81).
The
reduced
faster
than the disulfide. Addition of CO then furnished the carbonmonoxy complex 80.
1
H-NMR
of
the
CO
complexes
indicated
that
the
sulfide
underwent
intramolecular
binding without appreciable dimer formation.
UV/visible
band
spectroscopy
(384/460
or
in
DMSO
363/446
nm)
solution
which
or
was
aqueous
similar
suspension
to
the
hyper
showed
a
split
spectrum
of
carbonylated
Soret
cytochrome P450.
A
and
series
Groh
of
alkyl
and
28)82.
(Scheme
aryl
The
(oaminophenyl)triphenylporphyrin
(83).
Since
the
chain
introduced
by
detritylation
(HgIIZH2S)
indicated
that
spectra,
in
species.
The
was
first
he
alkyl
with
gave
chain
was
similar
introduction
of
CO
has
acid
derivatives
were
the
porphyrin
TrS-K+
thiol.
flexible
to
does
to
to
those
hold
of
lead
to
more
give
after
the
by
by
Collman
treating
difficult
85
and
to
insertion,
at
planar
formation
the
monochloride
obtain,
the
Deacetylation
iron
mercaptan
square
the
prepared
directly
S-tritylthiohexanoyl
AcS-K+.
or
However,
been
prepared
or
to
too
be
S-acetyl
free
were
may
wi h
either
the
porphyrins
chain
pentanoic
attached
treatment
toluene,
alkyl
(57)
S-protected
bromoalkyl
"mercaptan-tail"
C6
thio
(MeOH/NH3)
visible
metal
site;
six-coordinate
or
spectra
four-coordinate
of
the
group
the
iron(II)
low-spin
FeII-CO complexes.
To
ensure
CH3)
or
case
the
quen ly
greater
rigidity,
tails
(m-mercaptophenyl)acetic
potential
cleaved
thiol
with
was
sodium
derived
acid
introduced
87
as
borohydride
from
were
o-mercaptobenzoic
acid
attached
aminoporphyrin.
the
disulfide
to
give
the
to
(see
free
the
86
or
aryl
87)
(86;
which
X
was
"mercaptan-tail"
rins 90 and 91. As in the alkyl case, the aryl iron(II) species did not show five-coordina-
=
H
In
or
this
subseporphy-
139
140
tion.
Furthermore,
depending
on
addition
the
of
nature
of
CO
the
gave
mixtures
mercaptan
and
of
the
five-
and
temperature,
six-coordinate
suggesting
species,
a
tail-off/tail-
on equilibrium.
Deprotonation
of
the
mercaptan
to
give
the
mercaptide
was
attempted.
incomplete
deprotonation
The
extent
of
mercaptide formation depended both on the nature of the base
and
of
for
the
the
was
clean
system
tail.
Indeed,
for
most
tails
only
tail
system
(m-mercaptophenyl)-acetamide
and
gave
complete
a
using
six-coordinate
acetanilide
anion
92,
as
iron(II)-mercaptide-CO
deprotonation
base.
In
complex
the
93
occurred.
to
presence
whose
However
the
mercaptide
of
CO
visible
this
spectrum
exhibited a split Soret absorp ion at 450 and 380 nm, typical of cytochrome P450.
In
for
Sect.
3.A.
we
75
cytochrome
attaching
the
c .
thioether
(Scheme
allowed
95a-c,
29).
the
displacement
57
After
sulfide
model for cytochrome c.
ligand
the
iron
by
the
the
of
pyridine
FeII(C5Im)(TPP)(THT)
of
Rauchfuss
and
ligand
or
adopted
porphyrin
corresponding
thioether,
insertion
ordering
synthesis
and
to
with
containing
following
of
to
Buckingham
phenyl)triphenylporphyrin
porphyrins
referred
sulfoxide,
reduction,
affinities:
imidazole
the
periphery83.
anhydrides
or
Reaction
94
sulfone
precluded
>
R2S
this
as
a
model
strategy
of
afforded
groups
spectrophotometric
R2SO
(59)
alternative
titration
>
R2SO2.
system
as
of
(o-aminothe
tailed
in
60-90%
with
base
The
an
easy
effective
141
Smith
and
Bisset
model84,
P450
but
octalkylporphyrin.
placement
A
at
the
anediol
the
gave
hydrolysis
nately,
of
phyrins
ether
with
with
was
99
dithiols
tetrahydrofuran
no
the
yielded
containing
is
leading
sign
with
of
by
the
only
the
excess
a
to
give
RS—FeII-CO
melt
30).
the
disulfide
spectrum
of
sodium
of
of
suitably
Conversion
to
salt
he
98,
99.
Unfortufrom
the
acetoxymethylpor-
Treatment
hydride
an
dis-
1,6-hex-
100
l-(3-aminopropyl)imidazole
of
of
with
thiouronium
treatment
potential
nucleophilic
(Scheme
the
a
position
introduction
a
meso-methylporphyrins.
suspension
meso
to
in
formation
Alternatively,
with
synthesize
the
ready
96
afforded
oxidation
characteristic
to
to
susceptible
to
dimer
thiourea
unsuccessful.
octaethylporphyrin
approach
attached
substituent
accompanied
were
heme
were
acetoxymethylporphyrin
refluxing
generate
chelated
atom
the
97
by
to
complex
the
substituents
carbon
Heating
which
acetoxymethyl
ing
the
followed
attempts
disulfide
used
case
"benzylic"
chains.
bromide
also
this
meso-acetoxymethyl
functionalized
the
have
in
21
provided
of
meso-
in
reflux-
the
meso-che-
lated imidazole porphyrin 101 (Scheme 30), a potential model for T-state hemoglobin.
The success of the chelated heme approach to model heme proteins is due to its ability
to
control
captide,
tion
and
strongly
binding
the
coordination
thioether,
covalent
enhances
binding
ability.
binding
ligands
Addition
e.g.
of
of
a
metalloporphyrin.
attachment
without
the
imidazole,
one
to
the
need
of
pyridine,
equivalent
of
For
porphyrin
poorly
binding
increases
excess
external
chelation
can
base
mixture of four- and six-coordinate species, since K2 > K1 in Eq. 22.
to
an
ligands
the
ligand.
be
iron
e.g.
local
In
used
porphyrin
the
to
mer-
concentracase
lower
results
of
their
in
a
142
However
covalent
equivalent
of
attachment
base
which
of
the
can
base
bind
to
the
porphyrin
intramolecularly
to
give
provides
the
a
stoichiometric
desired
five-coordinate
species, provided dimerization is not significant.
Such
an
model
approach
has
porphyrins
been
e.g.
followed
by
picket-fence,
many
capped,
groups
to
produce
cyclophane
and
an
array
crowned,
of
different
strapped
and
basket-handle systems.
3.C. Porphyrins with Covalently Attached Quinone Groups
An
approach,
many
have
similar
researchers
to
to
stimulated
that
interest
photosynthesis,
of
prepare
where
as
the
chelated
porphyrins
possible
heme
with
models
photoinduced
for
charge
model
systems,
appended
the
transfer
has
quinone
primary
occurs
been
groups.
electron
from
adopted
Such
transfer
excited
by
systems
event
singlet
of
state
chlorophyll donors to nearby quinone acceptors.
One
of
the
earliest
such
models
dime hoxybenzene
103
was
(102)85.
(p-carboxyphenyl)tritolylporphyrin
followed
by
that
of
Kong
and
Condensation
demethylation
Loach
with
and
who
a
prepared
suitably
oxidation
a
meso-
substituted
furnished
he
desired
porphyrin-quinone pair 104 (Scheme 31).
A
et
similar
al.86-87*,
linkage
(ester
compounds,
transfer
series
who
of
varied
or
amide).
especially
occurred
substituted
both
105
from
the
EPR
(a,
tritolylporphyrins
length of
b;
porphyrin
and
n
to
=
105
the chain (n
laser
flash
3),
could
quinone;
have
=
2,
photolysis
adopt
subsequent
a
been
3,
prepared
4)
and the
studies
indicated
conformation
flipping
of
extended conformation could then prevent recombination and lead to a long-lived radical
by
Mcintosh
nature
of
that
the
these
in
which
electron
the
chain
to
an
143
ion
pair.
quinone
Wang
rings
al.88',
et
attached
lar
porphyrins
with
of
compound
have
prepared
to
an
the
a
appended
been
series
porphyrin
of
porphyrins
periphery
carotenoid
incorporated
by
group
into
106,107
amide
were
bilayer
also
lipid
having
linkages
either
one
(Scheme
prepared,
membranes.
and
or
32).
both
two
Simi-
classes
Photoconductivity
in
such membranes was enhanced relative to simple membranes89.
The
lead
to
Possible
use
of
amide
complications
solutions
or
as
to
ester
both
the
linkages
the
to
join
the
separation
and
orientation
problems
of
porphyrin
of
porphyrin-quinone
and
the
quinone
two
orientation
moieties
centers
and
can
of
can
vary.
prolonging
the change-separated species have included:
(i)
the
use
employing
of
more
capped,
rigid
strapped
spacers
and
to
doubly
separate
the
strapped
quinones
porphyrin
are
and
quinone
described
later
(models
in
the
appropriate sections);
(ii)
attempts
to
the porphyrin.
stabilize
the
transient
radical
ion
pair
by
introducing
other
groups
onto
144
Tabushi
chloride
in
et
(108),
which
the
al.
have
which,
after
reacted
selective
porphyrin-quinone
observed
but
no
mechanism
extended
and
the
relative
tion
of
TPPBQ
the
tively).
(40%)
110
The
constants
fixed
Illb
2-aldehyde
with
and
distances
charge
oxida ion,
fixed.
Efficient
The
in
benzaldehyde
TPPNO
photoinduced
is
were
and
Hlc
prepared
and
at
10,
(15%
10.5
and
recombination
for
have been reported92. The condensation of a linear tetrapyrrole 112 and an aldehyde to
has
33)
was
been
34)91.
(Scheme
yielded
benzaldehyde
(Scheme
quenching
distance
llla-c
benzoquinone
were
2,5-diacetoxybenzoyl
109
fluorescence
pyrrole
with
estimated
separation
with
furnished
porphyrin-quinone
and
TPPAO
57
compounds
reaction
pyrrole
TPP
and
proposed.
Diels-Alder
porphyrin-quinone
for
was
(25%).
111a.
corresponding
distance
orientations
2-anthraldehyde,
triphenylporphyrin
mono-o-amino
hydrolysis
Reac-
meso-(2-anthracenyl)
gave
by
the
product
condensation
and
11
18%
A.
these
of
respecThe
rate
compounds
145
form
mesosubstituted
113-115
The
(Scheme
bicyclooctane
Preliminary
porphyrins
35)
with
units
fluorescence
was
used
increasing
eliminate
yields
of
to
prepare
porphyrin-quinone
flexibility
the
free
and
base
another
series
distance
only
and
(6,
rotational
zinc
of
10
compounds
and
freedom
porphyrins
)93.
Å
14
is
indicate
allowed.
an
incre-
mental effect of distance on photochemical electron transfer93'.
A
similar
systems
incremental
shown
substituted
showed
in
which
of
36
which
were
116
(as
the
nickel
reduction
118
an
demonstrate
effect
Scheme
porphyrin
Deme hylation,
porphyrins
in
and
inverse
a
the
the
b.
double
The
exponential
multistep
pair
of
119a,
of
porphyrin-quinone
electron
quinone
of
dependence
transfer
separa ion
by
complex)
bonds
rate
rings
prepared
and
with
then
photoinduced
on
the
provide
the
Wittig
observed
of
of
phosphorus
furnished
electron
length
redox
the
oxidation
bis-quinone
a
was
condensation
transfer
the
porphyrin
potential
stabilize charge separation. Comparison with the mono-quinone etioporphyrin 119a
11794).
free
such
chain95.
In
120
was
gradient
and
the
meso-
ylide
the
in
with
the
base
systems
order
to
prepared96
may
thus
146
showed
the
approximately
decay
is
much
for
the
time
was
longer
than
generation
exponential
much
for
of
120
charge
group
121.
recombination
to
A
of
for
charge
Similarly
he
prepare
a
inhibited
by
state
of
states.
Moore
system
charge
transfer
photoinduced
importance
charge-separated
photodriven
was
the
120.
demonstrating
long-lived
aminophenyl)-bistolylporphyrin
carotenoid
decay
longer
the
electron
.
.
a
quinone
was
the
in
quinone
used
both
from
cases,
al.97,
complex
transfer
both
transfer
second
et
incorporating
separated
in
charge
but
119b
in
120
5,15-bis(4and
observed
carotenoid
a
since
to
the
porphyrin cation radical, to give a long-lived (us scale) C+ -P-Q - species.
3.D. Porphyrins with Covalently Attached Interactive Groups
A
number
than
of
o her
potential
Krishnan98
and
variable
tion
have
length
between
"tailed"
ligands.
prepared
122.
the
porphyrins
Using
Both
porphyrin
have
a
series
of
fluorescence
ring
been
mono-(m-
and
which
contain
groups
other
p-hydroxyphenyl)triphenylporphyrin,
derivatives
and
the
prepared
or
EPR
terminal
containing
data
a
phenoxyglycol
indicated
phenoxy
group,
chain
intramolecular
suggesting
Maiya
that
of
interacthe
tail
existed in a folded-over conformation.
Similarly
a
While
123
(65)
mono-(o-hydroxyphenyl)triphenylporphyrin
cyclodextrin
did
unit,
not
in
the
display
hope
the
of
preparing
desired
a
properties,
was
covalently
water-soluble
guest
moiety appears to induce novel conforma ional changes in aqueous solutions.
oxygen
inclusion
in
attached
carrying
the
to
model99.
cyclodextrin
147
Other
cyclodextrin
thylimidazoles
capped
included
in
hemes
an
have
prepared101
been
a-cyclodextrin
which
formed
using
a
1-substituted
pentacoordinated
2-mecomplex
with protoheme.
The
by
ability
several
Fe11
of
groups
porphyrins
to
attachment
of
a
suitable
DNA
the
helical
surface
where
DNA
Lown
and
Joshua101
124a-c
which
series
of
and
mimic
in
intercalator
reactive
prepared
the
properties
which
125,
a
generate
systems
have
compounds
protohemin,
to
prepare
a
suitable
to
iron
oxygen
the
of
could
might
give
deuterohemins
glycopeptide
from
is
radicals
bound
has
antitumour
porphyrin
species
derived
intercalator
oxygen
potential
an
series
of
126,
reactive
with
with
antibiotic
been
exploited
properties.
deliver
rise
an
to
Covalent
the
heme
to
DNA
scission.
attached
acridine
bleomycin.
A
similar
mono-p-aminophenyl)-tritotyIporphyrin
to
the
porphyrin
chain, has also been prepared which exhibits oxygen-dependent DNA cleaving ability102.
via
a
spermine
148
4. "Picket-Fence" Porphyrins and Related Species
4.A. "Picket-Fence" Porphyrins
Perhaps
the
porphyrin
of
most
successful
Collman.
of
Steric
the
heme
encumbrance
protein
about
active
the
site
metal
models
is
the
"picket-fence"
site
of
these
substituted
conformation
in
which
the
TPP
molecules depends on two factors:
(i)
due
to
steric
phenyl
repulsion,
rings
are
the
TPP
essentially
will
adopt
perpendicular
a
to
the
porphyrin
ring;
four
meso-
substituents
at
the
orr/io-positions of the phenyl rings will lie above and below the porphyrin plane, and
(ii)
for
TPP
molecules
depending
on
containing
the
bulk
mono-ortho-substituted
of
the
subs ituent,
phenyl
rings,
interconversion
of
separation
the
four
and,
possible
atropisomers may be achieved.
Collman
four
"protected
could
of
reasoned
pivalamido
pocket".
not
ligand.
bered
and
a
stable
since
syn hesis
located
Ligands
penetrate
excess
that
groups
the
The
e.g.
bulky
of
the
pocket,
a
pivalamido
the
porphyrin
ring
would
bind
to
the
metal
the
open
ensuring
could
five-coordination
molecule
form.
groups
tetraphenylporphyrin
of
dioxygen
complex
iron(II)
side
could
thereby
smaller
substituted
same
imidazole,
much
six-coordinate
the
on
would
This
should
on
even
not
oxygenated
prevent
be
in
face
the
sterically
complex
irreversible
having
give
presence
encum-
should
oxidation
a
but
be
through
close approach of two porphyrins and formation of a u-peroxo complex.
Condensation
acetic
acid
chloride
atropisomers
of
which
of
pyrrole
gave
to
the
(4a,
was
(55)
and
four
equivalents
meso-tetra(o-nitrophenyl)porphyrin
128
meso-tetra(o-aminophenyl)porphyrin
3ab
the
2a2b)
desired
were
aaaa
separated
isomer
129.
by
of
which
o-nitrobenzaldehyde
was
reduced
37)103,104.
(Scheme
chromatography,
Interconversion
of
sufficiently slow at room temperature to afford clean separation. Refluxing the unwanted
the
the
by
(53)
The
slowest
in
stannous
four
moving
atropisomers
was
149
products
further
gave
in
toluene
isolation
the
for
of
20
he
"picket-fence"
frozen
by
the
bulky
zation
of
these
min
aacta
effected
isomer.
porphyrin
of
the
to
130,
Several
the
have
studies
on
made105-107'.
been
the
amino
a,a,a,a-H2(TpivPP)
substituents.
atropisomers
reequilibration
Reaction
statistical
groups
with
which
the
in
physical
Treatment
mixture
allowing
pivaloyl
chloride
configuration
properties
with
and
FeBr2,
is
isomeri-
followed
by
II
reduction with Cr(acac)2 gave Fe (a,a,a,a,-TpivPP) 131.
Although
addi ion
FeB2(a,a,a,a-TpivPP),
"picket-fence"
compounds
pyridine,
side
was
oxygen
amounts
of
it
strong
was
was
less
prepared
piperidine,
reversible
of
field
suspected
than
(B
that
=
ligands
that
on
Im,
behaviour
decomposi ion.
Indeed,
in
the
l-Melm,
low
spin
binding
"open"
benzene
side.
l-n-Bulm,
the
solution
oxygen
six-coordinate
constant
A
of
at
All
25
complexes,
the
series
l-trityllm,
tetrahydrofuran)104.
tetrahydrothiophene,
binding
gave
the
complexes,
base
of
4-t-BuIm,
of
°C,
these
without
Fe(TpivPP)
were stable for long periods (ty2 2-3 months) in solution provided 2-4 equivalents of axial
on
the
six-coordinate
l,2-Me2Im,
showed
appreciable
(N-RIm)(O2)
150
base
talline
were
present
dioxygen
to
protect
complexes
Fen(TpivPP)(I-MeIm)(O2)
132a
the
unshielded
could
be
(Fig.
2)104108,
face.
Furthermore,
obtained104.
The
Fe"(TpivPP)(2-MeIm)
analytically
crystal
•
and its dioxygen adduct109 (Fig. 4) have been determined. Further structural information
Fig. 2. Perspective view of Fen(TpivPP)(I-MeIm)O2 (132a). Adapted from Ref. 108
pure,
structures
EtOH
(Fig.
crysof
3)
151
Fig. 4. Perspective view of Fe(02)(TpivPP)(2-MeIm). The dioxygen and imidazole are disordered.
The disorder has been idealized and only one concentration is shown in this figure, which is adapted
from Ref. 109
152
was
obtained
I.R108,110,111
by
measurements82'108*.
also
The
studied112.
been
and
reversible
Reviews
Mossbauer
binding
on
of
spectroscopy
oxygen
oxygen
to
binding
to
and
cobalt
magnetic
complexes
picket
fence,
susceptibility
of
and
TpivPP
related
has
systems
have also appeared32-37 113-115).
Binding
of
determination
crystals
a
five-coordinate
state
tive
binding
(at
Collman
increasing
as
in
be
solu ion.
deoxygenated
under
vacuum
solid
Hill
Y/l
-
(log
these
two
O2
a
in
terms
of
imidazole
its
bound
of
molecules
in
the
induces
solid
sufficient
which
are
strain
the
the
towards
of
and
Similar
=
that
non-cooperacooperative
molecules'
dimensions
the
this
(B
of
porphyrin
ring.
As
change
in
molecular
a
conforma-
crystallite
to
induce
affinity
of
the
oxygen
the
O2
concluded
region
the
oxygenated
in
enhances
move
of
was
regions
intermediate
shrinking
give
cycles116.
it
two
However,
to
Fe(TpivPP)B
P0z)
showed
prevented
between
many
of
log
an
this
and
over
vs.
compounds
and
cycling
samples
Y
pressures)
of
This
oxidation
by
low
solid
in
irreversible
plot
side
re-oxygenated.
iron
the
porphyrin
heme
be
the
presumably
change
the
five-coordinate
demonstrated
for
and
rationalized
numbers
dimensions
the
"picket-fence"
the
could
observable
binding
high
the
to
could
was
From
oxygen
oxygenation
tional
no
l,2-Me2Im).
binding.
on
binding
which
oxygenation
solid
on
species
produced
reversible
2-MeIm,
base
oxygen
FeII(TpivPP)(I-MeIm)(O2)
of
vacuum
second
of
remaining
deoxy
sites. This behaviour is reminiscent of the cooperative oxygen binding of hemoglobin.
Various
sation
other
of
Collman
(136),
et
by
al.
fonyl
chloride
complexes
at
directed
quent
have
reaction
rous
oxygen
sterically
hindered
TPP
derivatives
have
meso-a,a,a,a-tetra(o-aminophenyl)porphyrin
134
into
both
C
This
the
cavity,
iso-phthaloyl
38).
Despite
exhibited
attributed
to
only
the
series
the
amide
of
of
the
TPP
bulky
133
bulky
the
conden-
acid
chlorides.
and
and
H2(TTOSPP)
p-toluenesul-
"picket-fence",
oxidation
protons
which
coordinated
derivatives
by
(135)
dichloride
irreversible
acidic
protona ion
similar
prepared
with
H2(TphthPP)
compounds,
with
(Scheme
allowing
A
the
(129)
compounds
was
oxidation104*.
heme
prepared
H2(TAmPP)
respectively
of
0
25
also
of
been
129
on
were
dioxygen
137a-g
have
the
fer-
exposure
to
presumably
and
conse-
been
syn he-
sized under identical conditions by Bogatskii et al.117-118.
The
reaction
yielded
be
the
separated
CH3I,
o-H2(TAmPP)
of
tetra-isonicotinamide
by
followed
(129)
TPP
chromatography.
by
anion
The
and
meta
deriva ives
a
exchange,
were
excess
statistical
to
yield
(138)
also
isonicotinic
mixture
isonicotinamide
isonicotinamidophenyl)porphyrin-tetracation
para
with
as
groups
the
and
chloride
hen
soluble
39).
reduction
hydrochloride
atropisomers
were
water
(Scheme
prepared
of
which
methylated
could
with
tetrakis-(N-methyl-
The
corresponding
potentials,
basicity
and
reactivity with metal ions of the isomers were compared119.
Ort/iosubstituted
choosing
suitable
TPP
derivatives
substituents
on
the
have
been
phenyl
used
rings.
For
as
binuclear
example,
ligand
treatment
H2(TAmPP) 129 with maleic anhydride gave the tetrakis(o-maleamoylphenyl)porphyrin
systems
of
by
a,a,a,a-
153
(139)
in
insertion
the
90%
yield
(Scheme
at
a
rate
much
copper
by
the
carboxylates
40).
faster
In
aqueous
than
for
holds
the
DMF
this
unsubstituted
metal
ion
porphyrin
porphyrins.
in
a
undergoes
Rapid
position
copper
complexation
favourable
for
of
rapid
intramolecular transfer to the porphyrin nucleus120'.
Binuclear
synthetic
porphyrins
models
for
capable
the
of
iron/copper
binding
site
iron
of
and
copper
cytochrome
have
oxidase.
consists of a tetraphenylporphyrin ring with a covalently attached tetrapyridine ligand
been
One
inves igated
such
model
as
141
154
system,
(140)
ring
and
claim
tion
obtained
and
copper
magnetic
that
of
the
the
nicotinamide
tions
to
be
by
treating
41)121.
(Scheme
The
a,a,a,a-H2(TAmPP)
mixed
coordinated
susceptibility
to
of
metal
the
four
the
complex
conditions
necessary
for
groups123.
Instead,
used
which
for
the
locks
the
introduction
with
excess
with
iron
inserted
nicotinamide
nicotinamide
groups,
(129)
compound
examined12'.
metal
insertion
these
"pickets"
of
into the porphyrin ring, without fear of isomerization.
groups
divalent
In
into
authors
into
prepared,
contrast,
this
have
place
and
was
nicotinic
into
more
first-row
porphyrin
the
EPR
and
cause
coordinated
allowing
trivalent
and
Elliott
complex
anhydride
the
Krebs
isomeriza-
Ru"
to
the
forcing
condi-
transition
metals
155
Iron
porphyrin
oxidants
is
catalysis
believed
have
attempted
chiral
"picket-fence"
strate
olefin
to
the
to
catalytic
the
a,b,a,b-H2(TAraPP)
high
very
yield
little
chosen
as
an
the
porphyrin
was
reacted
This
(95%).
selectivity
catalyst
core.
which
The
was
in
diacid
chiral
active
chiral
epoxidation
olefins
porphyrins
acid
of
9-31%).
form
a
(142),
efficient
of
and
various
Instead
large
of
the
and
iodosylarenes
Groves
using
of
suitably
(Scheme
olefins
relatively
with
binaphthyl
rigid
chiral
l,l'-binaphthyl-2,2'-dicarboxylic
followed
by
methanolysis
enantiomeric
observed for variously substituted styrenes and aliphatic olefins.
excesses
the
by
42).
143a
as
Meyens
substituted
of
prepared
porphyrin
the
and
approach
were
chloride
appendage
(%ee,
using
intermediate.
stereochemistry
chloride
-H2(TAmPP)
more
the
The
the
could
hydroxyla ion
iron-oxo
epoxidation
optically
the
obtained
a,b,a,b
143b
an
and
reac ive
control
as
However,
was
a
species124.
with
group
appendage
with
to
iron-oxo
(142)
epoxidation
via
asymmetric
porphyrins
(R)-2-phenylpropanamido
in
of
proceed
sub-
reacting
With
was
an
formed
iodosylbenzene
group
was
cavity
about
acid
(144)
and
iron
insertion.
of
20-50%
were
156
More
ence
ence
recently
of
of
HCl
acid
converted
to
Tabushi
H2-Colloidal
and
chlorides
its
al.125
et
as
epoxide
a
have
platinum
cytochrome
and
slow
used
supported
P450
on
mimic.
reduction
Felll(TpivPP)Cl
in
the
pres-
poly(vinylpyrrolidone)
in
he
pres-
oxygenated
Under
such
the
ferric
complex
faces
of
of
conditions
cyclohexene
followed
by
is
formation
of the dioxygen ferrous complex completes the catalytic cycle.
A
porphyrin
(Scheme
43;
with
bis-ortho
the
on
145
bearing
pickets
=
CH3)126.
However,
R'
substituted
phenyl
both
only
rings
so
two
it
the
opposite
is
unlikely
ring
has
meso-positions
that
this
been
prepared
are
compound
substituted
will
be
a
successful oxygen binding model.
4.B. "Tailed Picket-Fence" Porphyrins
While
the
direct
solid
state,
such
oxygenation
studies
prevent
six-coordination
base
necessary
is
complex
formation.)
to
in
in
the
ensure
To
of
five-coordinate
solution
presence
complete
control
were
not
of
FeII
excess
coordination
coordination,
"picket-fence"
possible
on
Collman
since
sterically
the
"open"
et
was
the
al.,
observable
"picket-fence"
unhindered
face
and
adopted
heme" approach. Dispensing with external ligand, the base was covalently attached to
in
the
could
not
base.
(Excess
prevent
the
(μ-oxo
"chelated
the
position
ortho-phenyl
intramolecular
binding
of
to
TPP,
the
and
porphyrin
so
constrained
metal.
The
into
other
a
three
position
promoting
meso-phenyl
rings
carry
the "pickets" necessary to prevent irreversible oxidation.
a,a,a,a-H2(TAmPP)
Treating
"3-picket"
tion
for
2
h
atropisomers
isomer
urea
ing
re-equilibrated
an
imidazole
to
yield
gave
(S
a
=
2)
iron(II)
diamaenetic
free
=
H-NMR
(Momenteau.
0)
but
148
on
complex
not
1:1
of
confirmed
metal
the
the
the
observed,
presumably
had
observed
such
(3
a-
amide
of
using
"tail
proposed
and
chains
insertion
temperature
a
he
solu-
unwanted
Using
by
iron(II)
were
Travlor.
of
(The
147
gave
benzene
b-product.)
five-coordinate
decreasing
chloride
in
mixture
the
Direct
the
spectra
but
a
P-aminoporphyrin
149.
of
pivaloyl
Refluxing
chromatography.
yield
he
and
to
by
the
to
yields
formulation,
(S
increase
of
44).
group
separated
attached
porphyrins
1
equivalent
(Scheme
aminophenyl
to
was
3.2
(35%)
were
quantitative
15174.
e.g.
Drocess.
the
nearly
with
146
which
be
porphyrins,
to
the
147
could
leng h
spin
equilibrated
146,
linkages
FeBr2
(129)
a-aminophenylporphyrin
or
vary-
anhydrous
picket-fence"
five-coordinate
high
25
due
(due
°C)
to
dimerization
peaks
a
dimerization
in
the
chelated
heme systems.)
Addition
the
of
oxygen
expected
peak
pattern
plexes,
to
spectra
for
the
presumably
solutions
of
a
"pickets"
towards
of
the
high
diamagnetic
the
suggests
open
spin
compound
that
side
the
of
five-coordinate
for
the
oxygen
the
may
pocket,
iron(II)
oxygenated
a
be
compounds
species
ordered
suggestion
in
gave
151.
The
these
which
is
com-
awaiting
confirmation by X-ray crystallography.
A
similar
pyridine
series
covalently
of
"tailed
attached
to
picket-fence"
the
porphyrins
porphyrin
150
has
via
urea
periphery
been
synthesized
linkages127.
O2
with
a
and
CO
of
alkyl
binding to both series of porphyrins has been carried outl27_l29.
The
and
Sect.
use
aryl
meso-(0-aminophenyl)triphenylporphyrin
of
mercaptan-tail
3.B.
A
similar
porphyrins
series
of
tripivalamide-|3-aminophenylporphyrin
fence"
porphyrins
thioether
dioxygen74.
chain
has
154
also
as
cytochrome
compounds
147
to
(Scheme
45)82.
been
prepared
has
give
A
and
similar
is
57
P450
been
the
to
prepare
models
prepared
alkyl
series
been
by
155
capable
described
acylation
mercaptan
compound
reportedly
a
has
"tailed
with
of
an
reversibly
of
in
the
picketappended
binding
5. "Capped" Porphyrins and Related Species
5.A. "Capped" Porphyrins
The
direct
rins
was
condensation
exploited
molecules
a
by
benzene
of
aromatic
Baldwin
ring
and
was
aldehydes
co-workers
covalently
and
to
attached
pyrrole
prepare
to
all
to
form
"capped"
four
tetraphenylporphy-
porphyrins.
or//io-positions
of
In
these
the
meso-
phenyl rings, enclosing a volume of space above one face of the porphyrin ring. If the cap
was
sufficiently
prevented
nate
the
species.
On
under
was
tight
on
the
cap,
recognized
the
the
that
attempts
would
probably
to
"cap"
rin
"capped"
ring
to
porphyrin.
to
result
give
bases
the
provide
Unlike
on
in
low
very
alkylimidazoles,
open
barrier
benzene
tetraaldehyde
face
dioxygen
physical
a
the
(e.g.
the
smaller
a
condense
a
is the last step of the
not required.
of
binding
hand,
should
attached
the
face;
other
which
linkages
the
binding
enclosed
yields.
which
"picket-fence"
ring
result
in
molecule
would
be
to
[x-oxo
with
Instead
was
pyridine)
would
a
complex
porphyrin
the
condensed
porphyrin,
cyclization
able
to
fit
It
four
ester
units
were
pyrrole
of
synthesis, so chromatographic separation of
be
five-coordi-
formation.
by
necessary
with
should
a
the
to
give
porphy-
atropisomers
is
The
required
with
bromoethanol
159.
Reaction
of
tetraaldehyde
to
the
yield
156
158,
tetraaldehyde
was
prepared
followed
with
by
pyrrole
by
alkylation
condensation
in
refluxing
of
with
salicylaldehyde
pyromellitoyl
propionic
acid
(157)
chloride
yielded
the
"capped" porphyrin 160 after chromatographic purification (Scheme 46) (Fig. 5)130,131.
Fig. 5. Two perspective views of the "capped" porphyrin 160 as the free base. Adapted from
Ref. 140
The
same
sponding
reaction
sequence
"homologous"
or
using
"C3-capped"
2-(3-hydroxypropoxy)benzaldehyde
porphyrin
161
in
which
yielded
there
is
an
the
corre-
extra
methy-
lene group in each link of the cap131. In the latter case the yield of the cycliza ion reaction
was
much
larger
cap.
lower
(5%),
probably
reflecting
the
extra
entropy
factors
To
provide
steric
hindrance
on
he
uncapped
face
required
a
to
form
the
"naphthyl-C2-capped"
porphyrin 165 was similarly prepared (Scheme 47) from 159 and 162 via 163 and 164131.
Inser ion
of
iron
crystalline
four-coordinate
containing
excess
axial
into
the
high
base
"C2-capped"
spin
the
iron(II)
porphyrin,
porphyrin
five-coordinate
heme
followed
166
was
by
(Scheme
formed
reduction,
46).
which
reversible dioxygen binding at 25 0C The stability of the dioxygen adduct depended on
was
In
gave
a
solutions
capable
of
the
nature
and
concentration
48130.
Scheme
axial
base,
that
they
Unlike
the
the
larger
size
were
small,
appeared
that
a
side
the
cap.
of
of
e g.,
second
of
the
axial
"C2-capped"
the
"C3-Cap"
propylamine.
base
Oxygen
could
binding
base
porphyrin
permitted
For
weakly
was
and
166
the
position
which
could
binding
intermediate
of
size
two
bases
coordinate
to
the
reported
to
occur,
still
of
iron,
the
equilibria
only
bind
axial
bases,
such
as
probably
giving
a
a
in
single
provided
l-Melm,
through
it
the
pseudo-seven-
coordinate complex132-134).
The
been
natural
O2,
CO
and
131 137,l38
studied
-
porphyrins
H2(NapC2-Cap)
.
NO
affinities
It
was
and
other
(165)
>
of
found
that
synthetic
H2(C2-Cap)
a
series
of
FeII
O2
affinities
models
e.g.,
(160)
>
and
were
the
Fe"
CoII
capped
much
lower
complexes
H2(C2-CapNO2)
(161). In contrast CO bound more quickly than O2, and the rate of binding was indepen-
(167)
porphyrins
than
of
>
have
those
of
H2(TAP)
>
H2(C3-Cap)
Fig. 6. Perspective view OfFe111Cl(C2-Cap). Adapted from Ref. 141
dent
of
the
cap
in
terms
ra ionalized
H2(C2-Cap)
(160)
cap-porphyrin
more
of
expanded
too
must
the
might
of
to
the
unhindered
cap.
FeIIICl(C2-Cap)
small
exist
cap
be
effect
and
was
version
comparable
steric
5)140
(Fig.
under
system
and
a
separation
accommodated
Fe-O-O
size
in
argued
destabilized
to
the
(Fig.
6)14
either
solution.
by
Although
accommodate
against
That
a
a
porphyrins139.
model
crystal
indicated
CO
the
linear
Fe-C-O
"central"
steric
effect.
"peripheral"
steric
that
O2,
or
This
structures
the
a
system
of
both
phenyl
considerably
However,
effect
was
of
the
could
be
the
bent
methylene
chain linkages.
Studies142,143
used
to
NapC2-Cap
of
deduce
>
the
the
C2-Cap
paramagnetic
cap-porphyrin
>
C3-Cap,
shifts
in
separation.
which
1
the
The
H-NMR
relative
correlates
with
of
cavity
the
CoIICap
size
porphyrins
was
relative
in
oxygen
the
were
order
affinity
of
the iron(II) "capped" porphyrins.
Recently144
ified
the
"C2-capped"
"capped
porphyrin
porphyrin"
approach
in
a
which
has
pyridine
been
is
extended
covalently
to
bound
meso-aromatic rings of the parent "C2-capped" porphyrin, forming some kind of "strap"
prepare
to
two
a
mod-
opposite
(see
Chap.
6)
over
porphyrins
were
lowed
reduction.
by
functions
the
porphyrin
prepared
before
by
For
face
solubility
condensation
opposite
condensation
reasons
with
the
to
of
a
it
was
the
"cap".
Bis-amino
dinitro-tetraaldehyde
necessary
appropriate
3,5
to
pyridine
with
"C2-capped"
pyrrole
benzylate
diacid
the
folamino
compound.
Both
of the corresponding iron(II) C2-Capped strapped porphyrins 168a, b exhibit reversible
oxygen
binding
autoxidation
in
toluene
reactions
C4-Strapped
of
the
not
result
in
a
solution
complex
μ-oxo
at
C5-Strapped
(the
168a
complex,
room
complex
is
the
temperature
168b
several
strap
and
has
months).
cap
and
a
t½
show
of
good
several
Furthermore,
preventing
dimer
stability
days
while
decomposition
formation.
to
that
does
Although
a
kinetic study has not been made, it appears that the stability to autoxidation is due to the
low
affinity
that
of
of
iron(II)
introduction
of
the
iron(II)
C2-Capped
the
strap
complexes
complex
decreases
for
dioxygen.
(166)
the
(which
O2
Comparison
also
affinity
by
has
a
a
factor
of
low
of
P½
values
with
affinity)
shows
that
the
O2
4
for
C5-Strapped
the
complex 168b, and by a factor of 40 for the C4-Strapped complex 168a. This is due to a
combination
of
complex
in
porphyrin
upon
metal
offers
more
steric
tic
rings
a
steric
interactions.
domed
configuration
oxygenation.
further
resistance
interaction
preven ing
The
may
motion
to
the
presence
which
presence
occur
of
The
the
of
the
movement
between
pyridine
of
resists
the
the
cap
movement
strap
of
the
and
the
binding
the
metal
on
pyridine
towards
locks
of
the
more severe for the shorter C4-Strap, resulting in lower dioxygen affinity.
side-chains
porphyrin
the
metal
of
unoxygenated
towards
pyridine
oxygenation.
and
center.
the
This
to
the
the
Further-
meyo-aromawould
be
5.B. "Pocket" and "Tailed Pocket" Porphyrins
To
prepare
Collman
rin
is
a
et
approach
used
linked
to
to
system
which
discriminates
have
used
a
al.
to
prepare
provide
only
the
pocket
ide
can
steric
three
by
a
be
series
of
encumbrance
raera-phenyl
orientation
only
of
against
combina ion
of
at
one
the
face
of
leaving
bent
Fe-O-O
by
bending
of
binding
CO
"picket-fence"
porphyrins145.l46.
"pocket"
groups,
accommodated
the
the
the
an
unit
As
side.
toward
the
tilting
the
but
a
in
Oxygen
open
of
to
of
O2,
porphy-
benzene
this
may
side.
he
that
"capped"
above,
porphyrin,
open
and/or
relative
and
ring
case
it
bind
Carbon
is
within
monox-
linear
Fe-C-O
unit
one
equivalent
leading to decreased CO affinity.
Treatment
of
pivaloyl
chloride
with
benzene
a
a,a,a,a-H2(TAmPP)
formed
tris-acid
porphyrins
171
in
choice
acid
chloride
of
irreversible
oxidation.
good
A
the
chloride
yield
and
(129)
with
"mono-picket"
170
(>60%).
the
similar
sligh ly
porphyrin
under
The
high
dilution
volume
of
the
single
presence
of
strategy
was
more
169
employed
than
(Scheme
conditions
the
pocket
picket
to
49).
afforded
was
provided
prepare
porphyrins 172. However, in these compounds, the remaining ortho-ammo group is used
the
dictated
protection
the
of
Condensation
pocket
by
the
against
"tailed-pocket"
to attach the base, leaving no protection on the open face of the pocket. In contrast to the
"tailed
picket-fence"
porphyrins,
these
complexes
undergo
rapid
oxida ion
to
the
μ-oxo
complex145.
For
the
the
size
Piv)
iron(II)
of
derived
Although
Visible
from
171a
remained
medium
ranges
and
could
nant
species.
were
compared147.
O2
a
similar
he
model
two
coordination
and
coordinate
even
pockets
showed
within
which
CO
binding
of
the
O2
affinities
for
CO
systems,
affinity.
the
of
in
the
data
the
"pocket"
both
Since
systems
CO
iron(II)
of
excess
was
similar,
and
solvent
was
attributed
on
Fen(Poc-
that
base.
concen-
he
"picket-fence"
were
affinity
depended
six-coordination,
and
electronic
in
iron
showed
presence
increasing
five-coordinate
the
reduction
state
spectral
size
determined
reduced
MCD
five
and
While
showed
the
absorption
large
be
The
porphyrins
in
porphyrins,
pocket.
the
tration
"pocket"
the
domi-
porphyrins
the
"pocket"
effects
were
to
steric
the
hindrance of the cap which distorted the Fe-C-O unit from linearity.
“
5.C. Bis-Pocket" Porphyrins
Eventual
occurs
this,
steric
the
irreversible
because
of
encumbrance
both
is
octa-ort/io-substituted
TPP
bulk
substituents
for
porphyrin
molecules
but
stabilizing
the
FeII(P)
oxidation
steric
(where
the
ortho
ring.
The
would
(P2-)
complexes
pockets
form
oxygenated
a
barrier
the
tetrakis[2,4,6-tris(ethoxy)phenyl]porphyrin)
a
"picket-fence"
only
have
s ill
to
be
the
by
one
of
and
side
may
penetrated
be
Vaska
"capped"
the
By
by
approach
and
the
of
prepared.
pocket
close
Amundsen
dianion
on
been
protected
could
porphyrin.
is
the
present
choosing
formed
axial
of
two
have
porphyrins
molecule.
on
To
he
both
sides
bases
and
metal
centres,
prepared
the
avoid
correct
thus
hemes
tetrakis[2,4,6-tris(methoxy)phenyl]porphyrin
condensation
of
the
stituted benzaldehyde with pyrrole148. Balch has shown by 1H-NMR that although the
appropriate
of
diatomic
or
trisub-
or ho-methoxy
ature
substituents
proceeds
prepared
role
to
by
in
Suslick
refluxing
gave
sterically
hindered
was
However,
natural
base,
of
oxygen
systems,
to
porphyrin
species
173
is
reversible
very
observation
which
gave
at
room
hindered
1%
to
non-polar
other
to
the
pyr-
Metallation
and
of
iron(II)
complex
solvents
model
was
with
Addition
five-coordinate
in
attributed
yield.
temper-
complex
yield.
80%
a
compared
was
in
in
oxygenation
low
more
2,4,6-triphenylbenzaldehyde
the
1,2-dimethylimidazole,
affinity
an
obtain
oxidation
A
condensed
iron(II)
completely
formation,
Fe111(P)Cl11.
who
four-coordinate
capable
he
acid
complex
and
Fox149,
and
the
μ-oxo
FeIII(P)OH
propionic
reduction
which
prevent
form
at
0
30
complexes
non-polar
the
C
and
the
of
the
nature
binding site.
Covalent
to
attachment
prepare
by
porphyrin
Lindsey
separation
175
between
was
reaction
porphyrin
bases
by
(129).
The
after
porphyrin
gated
and
for
at
was
177.
rate
of
room
four
a
cofacial
fluoranil
of
for
constant
for
the
was
h.
and
then
transfer
of
Subsequent
from
yield
with
of
a,a,a,a-
the
of
in
the
Schiff
yield
furnished
to
base
"capped"
80-95%
compound
porphyrin
the
Schiff
reduction
176
this
used
adopted
where
the
insertion
of
be
was
quinone-tetraaldehyde
reacted
85%
metal
also
system
The
nature
reaction;
properties
electron
may
approach
.
porphyrin-quinone
groups
photochemical
TPP
an
10Å
which
24
desired
amino
be
intramolecular
yield
the
of
Such
porphyrin-quinone
to
174,
the
temperature
The
structure.
estimated
high
the
orr/iopositions
rigid
and
the
of
the
a
prepare
yielded
Acetylation
he
to
rings
reversibility
stirring
quinone
to
having
alkoxylation
NaBH3CN
50).
group
two
responsible
with
(Scheme
the
prepared
were
a
Mauzerall150
and
H2(TAmPP)
of
complexes
the
were
quinone
zinc
investi-
was
esti-
mated151.
6. Strapped Porphyrins
The
strapped
which
porphyrin
some
porphyrin
formed
group
macrocycle.
porphyrin,
cyclophane,
class
is
of
heme
protein
covalently
linked
to
The
thus
crowned,
usual
strategy
has
great
versatility
in
basket-handle,
embraces
corners
synthetic
allowing
pagoda,
models
two
been
he
etc.).
all
(usually
to
tie
types
The
those
diagonally
the
of
compounds
opposite)
strap
to
structures
porphyrins
may
be
in
of
an
a
already
made
(e.g.
singly-
or
doubly-strapped and may be classified according to the nature of the chain:
(i)
Simple
non-functionalized
alkyl
chains
whose
role
is
to
span
one
face
of
the
porphy-
rin, discouraging μ-oxo bridging and providing a more hydrophobic environment.
(ii)
Straps
incorporating
some
bulky
group
which
will
provide
more
steric
encumbrance
than a simple alkyl chain.
(iii)
Functionalized
interacting
with
five-coordina ion
straps
which
the
metal
or
to
incorporate
at
form
the
some
porphyrin
six-coordinate
group
core.
capable
These
mixed
may
ligand
of
be
binding
used
systems
to
L-M-L',
to
or
maintain
where
one ligand binds poorly to the metal.
6.A. Non-Functionalized Alkyl Straps
A
number
of
research
groups
have
reported
the
synthesis
of
simple
Ogoshi et al.152-154'condensed long-chain diamines with a difunctional etio-type porphy-
strapped
porphyrins.
rin
178
obtain
in
the
the
(Scheme
short
51).
used
at
0
C
same
(Scheme
ester
or
yield.
complexes
to
the
in
THF
ferric
porphyrin
model
to
iodosyl
benzene.
ligand
185a
models
by
investigate
prepared
in
cases
strain
0
the
into
an
Using
(shorter
185b.
A
to
served
similar
5-7
mimic
was
the
of
as
of
units
of
spectroscopy
Fe-CO
chains
by
Dieckmann
184
in
good
the
ferrous
irreversible
oxidation
strapped
a
cytochrome
P450
alkanes
substrate,
series
25%
amide-linked
as
he
with
in
or
unactivated
differentiation
Raman
and
rapid
an
por-
(180)
side
183,
for
al.135'
et
182
oxygenate
used
methylene
the
resonance
straps)
only
to
giving
ferrous
II
coupling
prepared
hydroxylation
itself
the
Battersby
carboxyl
to
that,
inhibited,
porphyrin
porphyrins
showed
claimed
mesoporphyrin
attempts
dilu ion
chromatography
was
to
bridged
oxidative
system
strap
was
strap
porphyrin
also
the
containing
attempt
C
have
high
after
complex.
of
strapped
20
156
instance
prepared
systems.
steric
the
sideways
at
Kuo
this
transformed
been
of
it
binding
the
intramolecular
under
yield
he
μ-oxo
chloride
give
porphyrin-catalyzed
some
also
natural
increased
In
the
being
has
were
the
In
and
the
to
move
acetone
under
oxygen
of
amide-linked
easily
Chang
53).
by
20-30%
spectroscopy
ligand
bis-acid
dilution
or
can
aqueous
(Scheme
the
triethylamine
in
observe
elaboration
ester-,
or
to
followed
strap
state.
185a
porphyrins
the
axial
forma ion
high
and
12)
absorption
bulky
rapid
under
6-10,
visible
Alternatively,
the
=
Attempts
in
formation,
chloroformate
(n
reacting
(181)
gave
of
species.
52).
Because
179
second
strategy,
amide
condensation
a
resulted
1,12-aminododecane
yield
isobutyl
basis
of
iron(II)
15
the
the
binding
five-coordinate
of
porphyrins
On
straps,
phyrins
presence
strapped
by
the
porphyrin
amide-linked
strapped
in
the
strap157.
O2
and
CO
displayed
correlation
between
a
stretching
and
Fe-C-O
Such
bending
vibrations has been observed158.
A
different
synthetic
porphyrins
where
Unlike
previous
of
the
the
porphyrin
intramolecular
sation
of
the
anion
186.
ylate
afforded
to
syntheses,
cyclization
187
unstable
to
and
of
Acid
he
"alkyl-strapped"
the
alkyl
In
strap
the
it
is
by
Baldwin
opposite
the
strap
In
the
final
step
the
strap
is
stretched
into
This
presence
not
of
is
prepared
with
immediately
190
able
in
to
excess
the
to
initially
position
which
condensed
23%
overall
enforce
base
the
prepare
rings
the
ring
is
(Scheme
gave
strapped
linked159,160.
are
and
porphyrin
benzyl
bis-dipyrromethane,
was
al.,
1,12-dibromododecane
condensation
porphyrin
was
which
with
et
meso-phenyl
ends.
chain-linked
189.
give
complex.
of
salicylaldehyde
examples
iron(II)
adopted
both
catalyzed
the
tetraacid
was
ortho-positions
attached
dialdehyde"
the
strategy
the
two
halves
formed
54).
he
by
Conden"strapped-
3,4-dimethylpyrrole-2-carboxafter
hydrogenolysis
with
yield.
trimethyl
As
five-coordination
six-coordinate
formed which did not bind oxygen. Reducing the concentration of base led to an increase
in
of
gave
orthoformate
the
the
species
previous
respective
191
was
172
173
of
the
four-coordinate
oxygenation
was
species
observed
at
which
-550C
underwent
similar
to
irreversible
unhindered
oxidation.
porphyrins,
While
warming
reversible
to
room
temperature caused μ-oxo bridge formation (Scheme 55).
A
a
similar
porphyrin
porphyrin.
would,
at
approach
with
very
Obviously
best,
give
in
was
short
this
very
used
by
alkyl
chains
case,
poor
et
al.161,162),
short
enough
Wijesekera
-
linking
yields.
opposite
Instead
the
who
to
corners
two
were
cause
attempting
deformation
of
a
preformed
halves
of
the
to
of
strap
the
porphyrin
porphyrin
were
assembled at each end of the strap, and only at the last step was the porphyrin 192 formed
by acid-catalyzed intramolecular cyclization under high dilution conditions (Scheme 56).
Visible
and
1
H-NMR
spectroscopy
and
X-ray
crystallography
(Fig.
7)
all
point
to
increas-
ing distortion of the ring as the length of the strap is decreased (192, n = 11, 10, 9). A
174
chain length of nine methylene units appears to be the lower limit; attempts to prepare
even
more
strained
porphyrins
with
shorter
straps
were
the metal complexes showed that the straps provided no steric protection.
Fig. 7. Ortep drawings of 192 (n = 9). Adapted from Ref. 162
unsuccessful.
Ligand
binding
to
175
6.B. Straps Containing Bulky Blocking Groups
Porphyrins
strapped
with
simple
alkyl
chains
are
poor
models
for
oxygen
binding
heme
proteins. In most cases the strap is too "floppy" and can be pushed to one side allowing \ioxo
bridge
leading
The
to
logical
formation.
oxygen
In
addition,
binding
extension
is
to
on
base
the
incorporate
is
not
open
prevented
face
some
bulky
and,
group
from
binding
consequently,
into
the
under
the
irreversible
strap
to
strap,
oxidation.
increase
the
steric encumbrance about one face.
One
of
the
initial
examples
of
the
strapped
porphyrin
approach
models was the cyclophane porphyrin 193 of Diekmann et al.l63). In this example steric
to
heme
protein
176
encumbrance
strap,
poor
was
porphyrin
yield
porphyrin
provided
cyclization
(5%
was
not
for
by
the
used
a
was
biphenyl
delayed
cyclization
as
a
heme
group
until
step
in
the
after
protein
the
strap.
final
step
repeated
model.
To
ensure
(Scheme
chromatographic
Instead
an
a
57).
fitting
Because
purification)
anthracene
across a preformed porphyrin (194) by means of amide linkages (Scheme 58). For the
tigh ly
was
of
this
strapped
177
anthracene-heme[6,6]cyclophane
~4.5Å
apart.
dione
(196)
of
second
a
A
gave
complexes
195
Diels-Alder
the
axial
"pagoda
base
198
two
porphyrin"
197
the
and
the
the
rings
possessing
even
homologous[
were
anthracene
anthracene
five-coordinate
198a
aromatic
on
underneath
being
heme[6,6]cyclophane
the
addition
an
ring
in
even
was
I M
7,7]
estimated
with
tighter
not
pocket.
observed,
I-MeIm.
compound
198b
to
be
l-phenyl-triazine-2,5-
The
were
Binding
the
iron(II)
anthracene-
used
to
study
the binding of isonitriles, CO and O2 within the pocket as models for the distal side steric
effects in heme proteins164_166.
The
Fe[6,6]
compared
effect),
This
to
and
due
to
between
steric
that
postulate
that
bent
heme
bulky
diatomic
reported
effects
the
differentiate
and
bending
large
Fe[7,7]
manifested
steric
to
a
( he
was
side
access
could
linear
showed
hemes
effect
distal
limited
compounds
198a
unhindered
this
suggested
but
to
cyclophane
flat
primarily
are
not
face.
in
due
isocyanide
or
of
the
to
bound
ligand
in
effects
led
CO
in
CO
the
could
Traylor
heme
and
only
a
rate167.
bound
state,
the
model
in
not
and
O2
small
association
steric
they
results
for
showed
repulsion
distal
ligands,
These
affinity
198b
in
While
molecules.
tilting
reduction
cyclophane
differen iate
his
proteins
colleagues
may
be
of
minor chemical significance.
Baldwin
benzene
capped"
which
did
adapted
ring
porphyrin
did
not
his
above
not
160,
prevent
prevent
strapped
one
u-oxo
face
the
porphyrin
absence
of
six-coordination
bridge
the
by
formation.
synthesis
59)160.
(Scheme
two
extra
ligands
An
to
prepare
Although
a
system
199
structurally
similar
to
linkages
such
even
as
more
resulted
I-MeIm
bulky
in
or
a
"floppy"
pyridine
strap,
with
the
and
a
"C2-
strap
which
incorporating
a
naphthalene ring as in 200, was no more successful.
Dolphin
durene
and
group
his
colleagues161,162)
protecting
one
face
have
(201)
prepared
(Scheme
a
series
60).
of
strapped
Incorporation
dard methods168 gave the corresponding heme complexes 202a-c. The crystal structure
of
porphyrins
iron
using
with
a
stan-
of
the
hemin
porphyrin
that
chloride
some
porphyrin
derivative
derived
planarity169.
from
201c
is
from
The
distortion
exists
essentially
flat.
7/7-base
201c
is
analogs
201b
and
201a.
The
to
their
interaction
with
binding
respect
constants
Me2Im)
to
durene
series
the
closer
of
to
also
1
(Fig.
spectra
in
H-NMR
porphyrin
heme
showed
for
the
the
durene-5/5
suggest
plane
derivatives
with
8)
data
than
of
the
imidazoles,
1,5-dicyclohexylimidazole
unhindered
despite
he
202a
optical
in
considerable
free
base
free
base
that
the
the
durene
(DcIm)
side
of
the
four-coordinate
differences
in
the
porphyrin
plane
201b,
durene
tighter
and
hemes
CO,
of
while
the
in
the
and
-4/4
5/5
have
been
are
these imidazoles do not coordinate on the capped (or distal) side of the heme. The size of
similar
for
The
(1,2-
within
steric
7/7-
studied
O2114170.
and
1,2-dimethylimidazole
distortion;
the
indicate
moiety
capped
porphyrins
isocyanides,
distortion
porphyrins
the
reasons,
179
180
Fig. 8. A SNOOPI diagram (E. K. Davies, plotting rou ine, 1984, Chemical Crystallography
Laboratory, 9 Parks Road. Oxford. England) of the hemin chloride of 202a (50% probability
contours for all atoms; hydrogen atoms have been omitted for clarity; the dashed bonds are used to
distinguish between the "strap" and the porphyrin skeleton)
202
the
distal
(TMIC)
cavity
and
extremely
coordination
was
examined
t-butylisocyanide
restrictive
of
distal
either
(t-BuNC),
environment
isocyanide.
using
the
which
of
The
the
bulky
differ
in
durene-4/4
isocyanides,
their
system,
Fe"(durene-7/7)(DcIm)
tosylmethylisocyanide
spatial
requirements.
however,
complex
202c exhibits a reduced overall affinity for CO relative to simple flat, open hemes; this is
inhibited
obtained
The
the
from
181
manifested
steric
in
effect
a
depressed
as
a
result
association
of
the
rate
durene-cap.
for
CO,
The
durene-5/5
and
was
interpreted170
and
-4/4
as
systems
a
distal
derived
from
202a and 202b also show reduced CO affinities compared to open hemes, but this results
predominantly
from
Fe(P)(DcIm)CO
complexes
increased
dissociation
because
of
rates
the
for
proximal
CO
steric
from
strain
the
induced
six-coordinate
by
the
porphy-
rin plane distortion. The five-coordinate hemes 202a • B - 202c • B (B = DcIm or 1,2Me2Im)
bind
skeletal
distortion.
202b
B
•
relative
heme
O2
(or
to
reversibly
A
other
values)
less
within
O2
complexes
to
distal
environments.
within
the
distal
similar
•
-
202c
encumbered
This
hemes)
severely
B
is
binding
extents,
entirely
arises
B
show
proximal
with
the
■
polar
the
amide
of
and
porphyrin
relative
of
to
CO
five-coordinate
kco/k°2
ratios
functions
in
higher
concept
moiety
of
discrimination
Significantly,
the
Fe-O2
effect
B complex
steric
considerably
incorporate
consistent
stabilizing
negligible
202a
system.
all
that
a
by the
from
distorted
•
hemes
pocket
implying
affinity for CO
distorted
his
202a
relative
to
10-fold reduced
electronic
(M
their
interactions
increasing
the
affinity
used
strap
of
the heme toward O2, relative to CO114'170.
The
same
adamantane
phane.
the
The
strap
greatly
synthetic
group
the
reduced
differentiation
porphyrin;
rates.
between
(as
one
structure171
crystal
and
strategy
across
of
in
a
Scheme
of
the
free
base
O2
and
isocyanides
and
the
O2
58)
porphyrin
CO,
However
CO
shown
face
(Fig.
adamantane
binding.
For
to
9)
CO
the
to
to
a
bulky
l,3-adamantane[6,6]
showed
bind
strap
was
give
no
the
free
Fe"
displayed
binding
cavity
complex
no
constant
cyclobetween
203
significant
was
with
steric
controlled
by the association rate, dissociation not being increased by the steric effects of the strap.
In
Chap.
et
contrast
6.C.),
al.172,
to
a
the
ligand
singly
showed
no
strapped
evidence
behaviour
of
the
pyridine-[5,5]
of
either
pyridine
cyclophane
internal
or
straps
heme
external
of
204
257
and
prepared
iron-pyridine
259
(see
by
Traylor
complex
forma-
tion. Indeed even the binding of CO to 204 failed to induce pyridine ligation even though
the
strap
binding
in
this
of
CO
case
and
bases
induces
to
sufficient
iron(II)
strain
porphyrins
to
prevent
is
synergistic.
internal
binding
Obviously
and
the
shorter
compound
behaves more like the anthracene and adamantane cyclophanes 198 and 203. The
Fig. 9. Perspective view of a l,3-adamantane[6,6] cyclophane porphyrin. Adapted from Ref. 171
204
182
pyridine
heme
manifested
between
204
in
CO
displays
lower
and
O2
severe
association
binding
steric
rates.
than
other
hindrance
to
Furthermore
models,
both
204
the
O2
and
shows
lower
CO
a
binding
binding,
greater
ratio
which
is
differentiation
being
possibly
due to enhanced binding of O2 in the tight polar pocket114.
Dolphin
and
Morgan173
have
prepared
the
series
of
strapped
Fig. 10 and Scheme 61. As a consequence of postponing porphyrin ring formation (a
porphyrins
illustrated
in
183
Fig. 10. Ortep drawings of a strapped porphyrin containing a thioether linkage. Morgan, B.,
Dolphin, D., Einstein, F. W. B., Jones, T., manuscript in preparation
specific example is given in Scheme 61) till the later stages on the synthesis, (i) a range of
strap
(ii)
ester
lengths
are
hydrocarbon
linkages
available,
straps
and
Compounds
including
strap
been
have
may
(iii)
some
be
increased
thioethers
prepared
of
which
employed,
may
stability
is
to
any
obtained
(Fig.
10),
phenol
(Scheme
61)
as
and photosynthetic charge separation, respectively.
lead
precluding
205
potential
and
distortion
polar
due
to
quinone
models
of
effects
for
the
206
the
porphyrin,
due
to
hydrocarbon
groups174
cytochrome
and
amide
c,
or
strap.
in
the
catalase
184
6. C Straps Containing Interactive Groups
The incorporation of potential ligands into the porphyrin strap has three advantages,
(i) A stoichiometric amount of ligand is built into the system, ensuring five-coordination without the addition of external ligand. In the case of nitrogen bases, mixtures
of six- and four-coordinate complexes are avoided.
(H) For ligands which bind poorly to iron(II) (e.g. thiolate), coordination would be
favoured by constraining the ligand into a position suitable for binding to the metal,
(iii) Because the strap is fixed to the porphyrin and the ligand in two positions, complications due to ligand replacement or dissociation will be minimized.
The strapped-porphyrin approach is also useful for orientating other interactive
groups (e.g., metal binding sites, electron donor/acceptor groups) into specific geometries with respect to the porphyrin.
185
Battersby
substituted
et
corresponding
38%
yield
syntheses
(Scheme
al.
pyridine
pyridine
(Scheme
is
have
shown
63)177.
A
prepared
ligands175.
diol
207
62).
in
The
Fig.
suitably
a
Reac ion
gave
series
strapped
the
the
ester-linked
molecular
11176).
of
of
A
functionalized
porphyrin
structure
cytochrome
diol
211
porphyrins
bis-acid
pyridine
of
a
straps
crystalline
P450
was
model
reacted
bearing
chloride
180
208a-c
in
side-product
was
under
the bis-acid chloride of mesoporphyrin II (180) to give the strapped porphyrin 212 in 25%
prepared
high
variously
with
the
up
of
to
these
similarly
dilution
with
186
yield.
The
protected-sulfur
group
with
potassium
S-acetyl
group
the
iron(II)
species.
whose
visible
monoxy
450
was
On
13
The
213
After
cleaved
exposure
absorption
cytochrome
nm.
derivative
thioacetate.
with
to
CO
spectrum
P450
C-NMR
was
iron
obtained
insertion
dimsyl
a
spectrum
of
a
the
split
to
CO
of
to
produce
iron(II)
major
hyper
13
the
displacement
reduction
sodium
six-coordinate
reproduced
spectrum
by
and
Soret
tosyloxy
iron(II)
the
five-coordinate
species
214
characteristics
complex
the
the
of
band
with
also
supported
an
state,
was
formed
the
carbon-
intense
band
at
RS—FeII-CO
the
formulation.
Several
binding
formed
bind
binucleating
two
metal
in
by the addition of
a
transition
diaza-18-crown-6
ether
also
complexes,
face
strapped
ions
of
metal
ring)
some
bulky
Iigands
porphyrin
the
give
from
over
216 to
and
metal
face
of
a
five-coordinate
species.
capable
of
Chang178,
bis-acid chloride
215, may
IA
217
or
binding
the
are
of
group
1-triphenylmethylimidazole)
which
porphyrin
the
a
its
one
prepared,
"crowned"
e her
ring)
Apart
control
(e.g.,
been
The
porphyrin
64).
steric
to
have
proximity.
bis-amino crown
(in
(Scheme
exerts
the
a
ion
porphyrins
close
IIA
cation
capability
porphyrin.
For
(in
the
the
crown
the
iron(II)
bind
only
on
the
unhindered
Oxygen
may
then
bind
under
the
crown to give a reasonably stable oxygen species (t1/2 > 1 hr at 250C in DMA).
o
Recently,
he
another
condensation
65)179.
(Scheme
and
cationic
cations
rate
225b
The
of
Bimetallic
FeIII,
Cu",
(see
diamines
porphyrin
Scheme
corresponding
species.
e.g.
salts
crown-strapped
of
similar
with
metalloporphyrin
complexes
fluorescence
indicated
219
67)
were
quenching
attachment
of
217
is
a
has
bis-amino
potential
prepared
was
the
to
the
and,
observed.
perchlorate
been
prepared
crown
ether
host
for
for
at
anionic
paramagnetic
Studies
salt
both
with
the
by
218
the
guest
perchlo-
porphyrin
metal
site and complexation of the ammonium species at the crown ether.
The
et
combination
al.180,
routes
(Scheme
refluxing
acetic
dichloride
tively.
metals.
who
222
This
of
crown
have
66).
acid
provided
compound
e her
prepared
and
porphyrin
the
w«o-tetra(benzo-18-crown-6)porphyrin
Condensation
or
the
is
of
reac ion
of
tetra-crown
reported
to
has
also
been
4'-formylbenzo-18-crown-6
carried
220
tetra(3,4-dihydroxyphenyl)porphyrin
ether
form
porphyrin
223
complexes
in
with
4%
out
by
Bogatskii
223
wi h
(221)
by
two
pyrrole
in
with
the
and
10%
yield
transition
and
group
respecI,
II
The
ton
et
combination
of
al.1B1
macrotetracyclic
biphenyl-linked
pyridine
at
The
bis-crown
55
0
C
crown
ether
under
ether
224
high
and
porphyrin
cryptand
with
dilution
the
226
has
was
recently
been
prepared
by
di-p-nitrophenyl
conditions
(45-54%
ester
after
tion to the tetra-amine 227 was effected by treating the zinc complex with diborane
extended
by
condensa ion
porphyrin
225a,
chromatography).
Hamilof
the
b
in
Reduc-
188
followed
by
selective
substrate
diammonium
within
the
demetallation
salts,
cavity
(Scheme
binding:
+
H3N(CH2)nNH3+
does
67).
transition
indeed
This
metal
(n
occur
=
was
multisite
cations
8-10),
complexing
are
bound
within
evidenced
by
the
the
by
species
the
is
capable
porphyrin
and
central
cavity.
That
large
upfield
shifts
methylene protons due to the shielding effects of the porphyrin and biphenyl rings.
of
alkyl
binding
of
the
189
A
copper
mimic
the
binding
EPR
The
copper-binding
cine
228
the
in
high
strap was
attached
of
chloride
under
chromatography
porphyrin
iron(III)
and
and
investigated
a
by
has
the
was
covalently
to
of
the
one side
copper(II)
dilution
68).
strap
of
ion.
Gunter
give
The
et
ions
a
extent
al.183,
for
of
of
rather
with
metal
an
231
the
different
in
phthalylglybinding
XII
72%
two
similar Fe/Cu strapped porphyrin (Scheme 69). Condensation of the tetramethyldipyr-
to
bis-acid
after
into
the
high
spin
centers
was
a
metal
approach
site,
corners.
yield
introduced
containing
to
oxidaseI82
opposite
mesoporphyrin
complex
attempt
from
copper
than to
differentially
between
somewhat
in
cytochrome
obtainable
porphyrin
be
coupling
porphyrin
site
229,
ring
229
could
a
a
non-square-planar
strapped
dinuclear
used
a
porphyrin
sulfide
the
to
iron-copper
derivative
need
the
gave
Metal
to
attached
the
bis-thiazole
of
diaminothiazole
the
EPR.
a
Because
high
(Scheme
into
been
characteristics
strap
yield.
Condensation
(230)
site
spectral
prepare
a
190
romethane
232
wi h
o-nitrobenzaldehyde
nitrophenylporphyrin.
ers
were
by
chromatography.
The
chloride)
(234),
separated
Insertion
of
followed
reduction
2.6-pyridylbis(4'thia-5'-pentanoyl
235.
(53),
After
iron
and
copper
to
and
by
the
the
oxidation
amino
cc,a-isomer
and
afforded
233
was
yielded
introduction
the
derivative
of
finally
the
bridging
5,15-meso(ortho-
the
atropisomers
condensed
strapped
ligands
with
porphyrin
gave
species
of
the type 236, whose magne ic properties were investigated184.
As
a
esh185,186
model
have
for
photosynthetic
prepared
a
and
electron-transfer
quinone-capped
porphyrin.
tersby
group,
1.4-dialkoxybenzene
deriva ives
237
bis-acid
chloride
(180)
strapped
porphyrins
on
strap
length
hydroquinones
studies
the
to
(Scheme
which
on
the
metal
ion
give
were
magnesium
and
that
the
70).
oxidized
complexes
theretore
were
Deprotection
to
the
239
d
in
with
and
wi h
7
b
that
Sanders
approach
and
boron
239a,
suggest
quinone
the
reacted
238
quinones
c,
the
systems,
Using
the
porphyrin
Gan-
the
Bat-
mesoporphyrin
15%
yield
trichloride
with
and
of
lead
quinone
II
depending
afforded
dioxide.
carbonyl
chromophores
the
'H-NMR
binds
are
to
perpen-
dicular187.
Since
zinc
oxygen
is
ligands,
sponding
zinc
auenchine
is
exclusively
intramolecular
complexes
observed
parallel
approach
nesium
complexes
of
in
the
five-coordinate
binding
239e,f
the
where
in
base
and
chromophores
with
239a,b
is
the
porphyrins
the
especially
free
chromophores
239c.d
of
added
and
possible,
chromophores
239e,f
is
are
a
lessened
ligands
zinc
but
has
is
much
low
affinity
in
the
present.
complexes
reduced
perpendicular"*'.
for
corre-
Fluorescence
where
for
The
the
close,
mag-
quenching
is more efficient for the longer chain 239b. f than for the shorter chain 239a. e; because of
intramolecular
binding
of
the
chromophores
in
239c,d
the
quenching
efficiency
is
similar
for both chain lengths.
High
dilution
coupling
of
the
bipyridyl
diols
240
with
180
gave
porphyrins 241 (Scheme 71) in 40-50% yield189 191 Treatment with iodomethane fur-
the
bipyridyl
strapped
191
192
nished
the
donor
(porphyrin)
potential
~200
assumed
strapped
and
an
photosyn hetic
reduced
was
methylviologen
fold
that
model.
relative
efficient
porphyrins
electron
Indeed
to
of
The
of
an
close
(methylviologen
fluorescence
mixtures
trapping
242.
acceptor
emissions
methylviologen
excited
electron
proximity
of
dication)
makes
of
241
and
an
and
unstrapped
occurred
to
electron
this
242
a
were
porphyrin.
give
the
two
the
condensation
It
radical
cations of the connected chromophores.
Hamilton
mesoporphyrin
strapped
resulted
,
al.192,
et
II
porphyrin
in
have
diacid
245
also
chloride
244
with
(Scheme
72).
prepared
180
a
binuclear
with
the
Ru(byp)2Cl2
followed
by
The
proximity
of
close
complex
bipyridyl
by
diol
metal
the
243.
Reaction
insertion
into
metal
centers,
two
of
the
of
the
porphyrin
estimated
at
4Å is reflected in both the luminescence and electrochemical properties of the complex.
6.D. Doubly-Strapped Porphyrins
We have
oxo
already seen
bridge
face.
formation
Steric
systems
of
that sterically encumbered porphyrins
if
encumbrance
Amundsen
any
on
and
four-coordinate
both
faces
Vaska148
species
of
the
in
porphyrin,
Suslick149,
and
may still
solution
as
may
be
binds
in
the
prevent
susceptible to
oxygen
on
he
"bis-pocket"
this
a
μ
open
porphyrin
bimolecular
oxida-
tion pathway.
Momenteau
TPP
derivatives
Baldwin's
and
his
having
"capped"
aldehyde
was
derivatives
246a-e
colleagues
two
and
"strapped"
reacted
to
straps
give
with
a
have
on
used
each
porphyrin
variety
chain-linked
a
combination
porphyrin
ring193.
syntheses159-160),
of
dialdehydes
dibromoalkyl
247.
The
of
In
approaches
a
the
strategy
sodium
and
TPP
by condensing the dialdehydes with pyrrole in refluxing propionic acid (Scheme 73).
to
prepare
reminiscent
salt
of
of
salicyl-
p-(dibromoalkyl)benzene
ring
was
then
formed
193
After
in
removal
low
minant
of
polymeric
overall
yield.
isomer.
To
formation
of
(Scheme
74).
The
the
increase
the
bridges
was
phenyl)porphyrin
DMF
at
three
isomers
adjacent
yield
of
(10%
the
delayed
(250).
100°C,
yield)
Alkyla ion
followed
obtained
product
more
until
248c
interesting
after
(249)
and
with
by
were
cis-linked
Tetra(o-methoxyphenyl)porphyrin
o-methoxybenzaldehyde
in
materials,
unwanted
the
the
dibromo
cross
to
to
the
pyrrole
provide
246
isomers 248. In this case the major product of each reaction was
and
tetra(o-hydroxy-
under
of
predoisomers,
condensation
from
isolation
the
trans-linked
obtained
derivatives
led
chromatography
often
porphyrin-forming
was
demethylated
chromatography
by
was
high
the
dilution
hree
porphyrin
he desired cross trans-
linked isomer 248a. The starting porphyrin 250 was used as a mixture of the four possible
atropisomers since the conditions of the condensation would lead to equilibra ion.
The
degree
tion
of
the
and
is
easily
of
iron
diatomic
molecules,
insertion
derivative
tion
the
under
O2
less
(1
The
In
contrast
chains
iron(II)
atm),
t1/2
do
is
the
other
not
trans-linked
of
rates
of
248c
has
isomers,
u-oxo
248a
complex.
faces
room
t½
Similarly,
oxida-
unhindered
are
hindered,
bases
temperature.
for
7-54
iron(II)
and
face
nitrogenous
at
to
six-coordinate
one
of
he
compared
metallation
both
ligation
oxidation
isomer
the
where
preventing
minutes
the
oxidation
the
isomer
irreversible
1.5-10.5
to
by
cis-linked
inhibit
isomers
for
illustrated
While
cross
FeIII(P)OH
hindered
is
adjacent
reluctantly.
the
four-coordinate
hematin
of
encumbrance
isomers.
metallated.
undergo
the
steric
various
oxidation
seconds
in
toluene
complex
to
for
the
oxida-
at
is
or
For
25
11-25
0
C
min
for the cross trans-linked isomer compared to 1.5-12 min for the other two isomers194.
The
basket-handle
metallation
try.
and
Detailed
paralleling
studies
the
and
binding
earlier
picket-fence
of
on
small
the
porphyrins
molecules
electrochemistry
electrochemical
studies
but
show
in
of
the
on
dramatic
their
the
redox
iron
effects
and
complex
free
base,
not
only
during
coordination
have
been
magnesium
chemismade195'
and
zinc
complexes of 192 and 201196.
A
similar
triglycolic
not
doubly-strapped
atropisomer
a,b,a,b
dichloride
cause
at
significant
of
room
porphyrin
has
been
reported
by
meso-tetra(o-aminophenyl)porphyrin
temperature
isomerization
of
in
the
the
presence
atropisomer.
251 was obtained in 32% yield after chromatography (Scheme 75).
(142)
of
Zhilina
was
pyridine,
The
et
al.
.
acylated
conditions
doubly-strapped
hat
The
with
do
porphyrin
In
a
contrast
one-step
units.
yielded
desired
the
synthesis
Thus,
a
to
sequential
to
prepare
condensation
mixture
cross
characteristic
of
of
trans-linked
a
the
oligomers
of
porphyrin
H-NMR
252
polymers,
obtained
spectrum.
bridging
straps,
sandwiched
bis-aldehyde
and
porphyrin,
1
symmetric
introduction
with
and
in
Weiser
between
pyrrole
three
in
two
parallel
refluxing
doubly-strapped
~0.1%,
was
Deme hylation
and
Staabl98
and
easily
used
quinone
propionic
acid
porphyrins.
The
identified
oxidation
by
then
its
yielded
the desired bis-quinone porphyrin 253 (Scheme 76).
A
further
doubly-strapped
globin,
of
the
refinement
models
incorporation
natural
of
system
to
he
production
containing
a
nitrogen
while
the
mimic the distal, oxygen-binding face.
different
base
steric
of
heme
straps.
into
one
protein
models
As
models
strap
would
encumbrance
provided
was
for
simulate
by
the
the
synthesis
hemoglobin
or
the
proximal
second
strap
of
myoface
would
Momenteau's
compounds
in
route
which
to
an
doubly-strapped
axial
base
tion
of
tetra(o-hydroxyphenyl)porphyrin
lent
of
1.12-dibromododecane
on
whether
mixture
adjacent
was
linked
isomer
257
porphyrin
259
case
straps
the
Following
with
iron
a
opposite
by
from
tied
the
insertion
of
of
and
to
tic
easily
one
four
two
(254)
preparative
prepared
were
(mixture
mixture
was
into
of
(5%
adapted
256
isomers)
overall
wi h
the
yield)
porphyrin
reduction,
skeleton
visible
by
were
(Scheme
amide
absorption
1
and
one
H-NMR
cross
77).
(142);
linkages
equivadepending
linked.
desired
a,b.a,(3-tetra(o-aminophenyl)porphyrin
produce
Condensa-
porphyrins,
groups
and
to
straps199'
the
singly-linked
meso-phenyl
3,5-bis(3-bromopropyl)pyridine
isolated
was
or
incorporated
(250)
gave
(255),
reacted
porphyrins
was
A
This
transsimilar
in
this
(Scheme
78).
spectra
of
both
compounds were consistent with a five-coordinate high spin (S = 2) iron(II) complex.
The
by
rate constants for the
laser
flash
photolysis.
The
association
O2
and
affinity
of
dissociation of O2 and CO were determined
the
"amide"
linked
system
was
higher
than
that of the "ether" linked compound p½ 18.6 vs 2 torr) as a result of a difference of a
factor
stability
of
ca.
of
10
the
in
he
"amide"
O2
dissociation
oxygenated
rates
species
(10~4
was
koffO2
attributed
4
vs
to
s_l).
0.5
the
This
presence
group and the possibility of hydrogen-bonding with the terminal oxygen atom. The low
increase
of
the
in
N-H
temperature
(-27°C)
quivalence of
1
H-NMR
spectrum
supported
he pyrrole protons as well as
this
hypothesis,
the
observed
ine-
he shifts of the amide protons suggesting a
preferred orientation of the oxygen molecule towards the amide N-H groups200.
To better model the hemoglobin and myoglobin active sites a doubly strapped heme
260 was prepared incorporating a pendant imidazole (Scheme 79)201. 260 was capable of
binding oxygen to give a rela ively stable oxygenated species (lifetime was about one day
in dry toluene under 1 atm O2). The kinetics of O2 and CO binding have been determined and initial comparisons with the comparable "pendant pyridine" porphyrins show:
(i)
O2 and CO combina ion rates are practically constant in the three pendant base
(ii)
a reduction in konO2 in the imidazole porphyrin due to a combination of hydrogen
porphyrins, and
bonding with the amide N-H and the greater basicity of imidazole over pyridine.
Comparison
of
the
pendant
imidazole
model
with
myoglobin
or
isolated
hemoglobin
chains shows that the model reacts 10 times faster with O2 and that the dissociation rate is
approximately 100 times faster than in the natural systems.
With the availability of the differentially protected coproporphyrin I 261, Battersby
and Hamilton adapted heir syntheses to the production of doubly-strapped porphyrins
80)202.
(Scheme
ridine
yielded
esters
and
263
to
with
acid
give
difficult
so
aqueous
temperature
an
CH2Cl2-
was
cycles
in
In
after
the
judged
to
by
264
the
species
compound
with
a
a
of
the
oxygenated
could
be
repeated
the
resistance
more
species
of
=
of
t1/2
the
on
2)
of
the
was
the
the
found
strap.
basis
of
five-coordinate.
accomplished
by
significant
min
CO-porphyrin
passage
complexes
visible
at
the
of
to
room
formed
into
irreversible
be
Reduction
the
was
CO
diol
to
Exposure
15
species
by passing
benzyl
anthracene
pyridine
oxygenated
without
unhindered
with
approximately
displaced
was
times
3,5-bis(3-hydroxypropyl)py-
insertion
of
which
(S
stable
could be
six
Iron
introduc ion
iron(II)
with
Hydrogenolysis
condensation
(27%).
spin
O2
261
(33%).
high
DMF
to
262
be
h at 20°C). The
contrast
chloride
followed
porphyrin
furnished
was
was
inserted
oxygenated
regeneration
O2-CO
bis-acid
porphyrin
formation
was
dithionite
in
the
doubly-bridged
metal
approximately 2
and
chloride
spectrum
gave
of
pyridine-strapped
the
the
absorption
oxygen
Reaction
the
(t1/2
solution,
O2.
Such
oxidation.
This
to
displacement
of CO by O2.
The
further
reported203'.
the
As
anthracene
esters
and
substituted
yield
of
significant
diol
81).
of
the
263
treatment
imidazole
(Scheme
cycles
refinement
before,
with
to
incorporating
differen ially
give
oxalyl
diol
266.
The
iron(II)
the
oxidation
imidazole
the
(by
occurred.
was
265
(57%).
bis-acid
capable
of
the
the
was
still
been
was
removal
was
267
of
the
with
oxygen
with
benzyl
the
obtained
being
oxygenated
recently
reacted
reacted
was
reversible
pressure)
for
t1/2
has
261
After
chloride
porphyrin
reducing
The
ligand
coproporphyrin
doubly-bridged
complex
oxygenation-deoxygenation
irreversible
porphyrin
chloride,
The
an
protected
in
binding,
possible
species
N22%
four
before
was
ca.
24 h at room temperature in DMF solution.
Recognizing
straps
(Scheme
imidazole
that
containing
81)203).
268b
with
the
a
pendant-imidazole
1,5-disubstituted
Coupling
he
bis-acid
strap
imidazole
1,5-bis(4-hydroxybutyl)
chloride
of
the
268
268a
somewhat
were
floppy,
prepared
or
anthracene-strapped
doubly-bridged systems 269a, b in 23% and 6% yield. Distortion of the porphyrin ring
more
as
rigid
before
l,5-bis(3-hydroxypropyl)porphyrin
gave
he
198
from
planarity
to
accommodate
the
shorter
strap
was
believed
responsible
for
the
low
yield of 269b and also for the lesser stability of the oxygenated iron(II) species. For 269a,
the
iron(II)
ture.
Ten
dation
was
complex
could
reversibly
oxygenation-deoxygenation
significant,
and
only
bind
oxygen
in
cycles
could
be
20%
irreversible
DMF
solution
performed
oxida ion
at
ambient
before
occurred
tempera-
irreversible
after
2
days
oxiin
solution.
Acknowledgements. This work was supported by the United States National Institute of Health
(AM 17989) and the Canadian Natural Sciences and Engineering Research Council.
7. List of Abbreviations
acac
AQ
bipy
1-n-Bulm
4-t-BuIm
t-BuNC
BO
CO
Cys
cyt
DCIM
DDO
DMA
acetylacetonate
Anthraquinone
bipyridyl
1-n-Butylimidazole
4-t-Butylimidazole
t-Butvlisocyanide
Benzoquinone
Carbon monoxide
Cysteine
cytochrome
1,5-Dicyclohexylimidazolc
2,3-Dichloro-5,6-dicyano-l,4benzoquinone
N1N-DimethyIacetamide
DMF
DMSO
DNA
ee
Et3N
EXAFS
N1N-DimethyIformamide
Dimethylsulfoxide
Deoxyribonucleic acid
enantiomeric excess
Triethylamine
Extended X-Ray Absorption
Fine Structure
H2(OEP)
Octaethylporphyrin
general porphyrin
H2(P)
H2(TAP)
Tetra(p-methoxyphenyl)porphyrin
H2(T(OH)PP) Tetra(o-hydroxyphcnyl)porphyrin
H2(TPP)
Tetraphenylporphyrin
H2(TTP)
His
Hm
Im
MCD
I-MeIm
l,2-Me2Im
MeOH
Met
NQ
Tetra(p-tolyl)porphyrin
Histidine
Heme
Imidazole
Magnetic Circular Dichroism
I-Methylimidazole
1,2-Dimethylimidazole
Methanol
Methionine
Naphthoquinone
phth
piv
THF
THT
TMIC
Tos
Tr
l-trityllm
Tyr
phthaloyl
pivalamido
Tetrahydrofuran
Tetrahydrothiophene
Tosylmethylisocyanide
Tosylate
Trityl
I-Tritylimidazole
Tyrosine
8. References
1.
2.
3.
4.
5.
Hemoglobin and Oxygen Binding (Ho, C, Ed.), Elsevier Biomedical, New York 1982
Mathews, F. S.: Prog. Biophys. Mol. Biol. 45, 1 (1985)
Hatefi, Y.: Ann. Rev. Biochem. 54, 1015 (1985)
Frew, J. E., Jones, P.: Adv. Inorg. Bioinorg. Mech. 3, 175 (1984)
Murray, R. T.. Fisher. M. T., Debrunner, P. G., Sligar. S. G.: Top. Mol. Struct. Biol. 6
(Metalloproteins, Pt. 1). 157 (1985)
6. The Porphyrins (Dolphin, D., Ed.), Academic Press, New York, Vol. VII, Part B, 1978
8. NATO Adv. Study Inst. Ser., Ser. C, 89 (The Biological Chemistry of Iron) (1981)
8. Kao, O. H. W., Wang, J. H.: Biochemistry 4, 342 (1965)
9. Hammond, G. S., Wu, C H. S.: Adv. Chem. Ser. 77, 186 (1968)
10. Cohen, I. A., Caughey, W. S.: Biochemistry 7, 636 (1968)
11. Latos-Grazynski. L., Cheng, R.-J., La Mar, G. N., Balch, A. L.: J. Am. Chem. Soc 104, 5992
(1982)
12. Holquist. D. E., Saunes, L. J., Juckett, D. A.: Curr. Top. Cell. Regul. 24, 287 (1984)
13. Brunori. M., Falcioni, G., Fioretti, E., Giardina, B., Rotilio, G.: Eur. J. Biochem. 53, 99
(1975)
14. Fee, J. A.: Metal Ion Activation of Dioxygen (Ed. Spiro, T. G.), Wiley Interscience. 209-237
(1980)
15. Phillips, S. E. V., Schoenborn, B. P.: Nature (London) 292, 81 (1981)
16. Shaanan, B.: Nature (London) 296, 683 (1982)
17. Perutz, M. F.: Scientific American 239{6), 68 (1978)
18. Brault, D., Rougee, M.: Biochemistry 13, 4591 (1974)
19. Brault, D., Rougee, M.: ibid. 13, 4598 (1974)
20. Scheidt, W. R., Reed, C A.: Chem. Rev. 81, 543 (1981)
21. Stynes, D. V., Stynes, H. C, James, B. R., Ibers, J. A.: J. Am. Chem. Soc 95, 1796 (1973)
22. Anderson, D. L., Weschler, C J., Basolo, F.: ibid. 96, 5599 (1974)
23. Almog, J., Baldwin, J. E., Dyer, R. L., Huff, J.. Wilkerson, C J.: ibid. 96, 5600 (1974)
24. Brinigar, W. S., Chang, C K.: ibid. 96, 5595 (1974)
25. Wagner, G. C, Kassner, R. J.: ibid. 96, 5593 (1974)
26. Traylor, T. G., Chang, C. K., Geibel, J., Berzinis, A.. Mincey, T., Cannon, G.: ibid. 101,
6716 (1979)
27. Gibson, Q. H.: Prog. Biophys., Biophys. Chem. 9, 1 (1959)
28. Basolo, F., Hoffman, B. M., Ibers, J. A.: Acc Chem. Res. 8, 384 (1975)
29. James, B. R.. Addison, A. W., Cairns, M.. Dolphin. D., Farreli, N. P.. Paulson, D. R.,
Walker, S.: Fundamental Research in Homogeneous Catalysis (Tsutsui, M.. Ed.), Plenum
Press: New York, Vol. 3, p. 751 (1979)
30. Wang, J. H.: Ace Chem. Res. 3, 90 (1970)
31. Leal, O., Anderson, D. L., Bowman, R. G., Basolo, F., Burwell, R. L.: J. Am. Chem. Soe
97, 5125 (1975)
32. Collman, J. P.: Ace Chem. Res. 10, 265 (1977)
33. Jones, R. D., Summerville, D. A., Basolo, F.: Chem. Rev. 79, 139 (1979)
34. Smith, P. D., James, B. R., Dolphin, D. H.: Coord. Chem. Rev. 39, 31 (1981)
200
36. Traylor, T. G.: Acc Chem. Res. 14, 102 (1981)
37. Bogatskii, A. V., Zhilina, Z. I.: Russ. Chem. Rev. 57, 592 (1982)
38. Collman, J. P., Halpert, T. R., Suslick, K. S.: Metal Ion Activation of Dioxygen (Spiro, T. G.,
Ed.), Wiley: New York, p. 1 (1980)
39. Lautsch, V. W., Wiemer, B., Zschenderlein, P., Kraege, H. J.. Bandel, W., Gunther, D.,
Schulz, G., Gnichtel, H.: Kolloid. Z. 161, 36 (1958)
40. Losse, G., Muller1 G.: Hoppe-Scyler's Z. Physiol. Chem. 327, 205 (1962)
41. Van der Heijden, A., Peer, H. G., Van den Oord, A. H. A.: J. Chem. Soc., Chem. Commun., 369 (1971)
42. Warme, P. K., Hager, L. P.: Biochemistry 9, 1599 (1970)
43. Momenteau, M., Rougee, M., Loock, B.: Eur. J. Biochem. 71, 63 (1976)
44. Castro, C E.: Bioinorg. Chem. 4, 45 (1974)
45. Molokoedov, A. S., Fillippovich, E. I., Mazakova, N. A., Evstigneeva, R. P.: Zhur. Obshch.
Khim. Ed. Engl. 47, 1070 (1977)
46. Kazakova, N. A., Radyukhin, V. A., Luzgiva, V. N., Filippovich, E. I., Kamyshan, N. V.,
Kudryavtseva, E. V., Evstigneeva, R. P.: ibid. 52, 1896 (1982)
47. Momenteau, M.. Loock, B.: Biochim. Biophys. Acta 343, 535 (1974)
48. Selve, C, Niedercorn, F.. Nacro, M., Castro, B., Gabriel, M.: Tetrahedron 37, 1893 (1981)
49. Selve, C, Niedercorn, F., Nacro, M., Castro, B., Gabriel, M.: ibid. 37, 1903 (1981)
50. Gabriel, M., Grange, J., Niedercorn. F., Selve, C, Castro, B.: ibid. 37, 1913 (1981)
51. Goulon, J., Goulon, C, Niedercorn. F.. Selve, C, Castro, B.: ibid. 37, 2707 (1981)
52. Chang. C K., Traylor, T. G.: Proc Natl. Acad. Sci. U.S.A. 70, 2647 (1973)
53. Chang, C K., Traylor, T. G.: J. Am. Chem. Soc 95, 5810 (1973)
54. Chang, C K., Traylor, T. G.: ibid. 95, 8475 (1973)
55. Brinigar, W. S., Chang, C K., Geibel, J., Traylor, T. G.: ibid. 96, 5597 (1974)
56. Geibel, J., Chang, C K., Traylor, T. G.: ibid. 97, 5924 (1975)
57. Dolphin, D., Hiom, J., Paine III, J. B.: Heterocycles 16, 417 (1981)
58. Traylor, T. G., Campbell, D., Sharma, V., Geibel, J.: J. Am. Chem. Soc 101, 5376 (1979)
59. Traylor, T. G., White, D. K., Campbell. D. H., Berzinis, A. P.: ibid. 103, 4932 (1981)
60. Traylor, T. G., Mitchell, M. J., Cicone, G. P., Nelson, S.: ibid. 104, 4986 (1982)
61. Traylor, T. G., Tatsuno, T., Powell, D. W., Cannon, J. B.: J. Chem. Soe Chem. Commun.,
732 (1977)
62. Traylor. T. G., Berzinis, A. P.: J. Am. Chem. Soe 102, 2844 (1980)
63. Tabushi, I., Sasaki, T.: Tetrahedron Lett. 23, 1913 (1982)
64. Tabushi, I., Kugimiyua, S., Kinnaird, M. G., Sasaki, T.: J. Am. Chem. Soe 107, 4192 (1985)
65. Tabushi, I., Kugimiya, S., Sasaki, T.: ibid. 107, 5159 (1985)
66. Denniss, J. S., Sanders, J. K. M.: Tetrahedron Lett., 295 (1978)
67. Boxer, S. G., Wright, K. A.: J. Am. Chem. Soe 101, 6791 (1979)
68. Momenteau, M., Loock, B., Bisagni, E.. Rougee, M.: Can. J. Chem. 57, 1804 (1979)
69. Callot, H. J.: Tetrahedron, 899 (1973)
70. Lavalette, D., Tetreau, C, Momenteau, M.: J. Am. Chem. Soe 101, 5395 (1979)
71. Lavalette, D., Momenteau, M.: J. Chem. Soe, Perkin Trans. 2, 385 (1982)
72. More, K. M., Eaton, S. S., Eaton, G. R.: Inorg. Chem. 20, 2641 (1981)
73. Damoder, R., More, K. M., Eaton, G. R., Eaton, S. S.: J. Am. Chem. Soe 105, 2147 (1983)
74. Damoder, R., More, K. M., Eaton, G. R., Eaton, S. S.: Inorg. Chem. 22, 2836 (1983)
75. Collman, J. P., Brauman, J. I., Doxsee, K. M., Halbert, T. R., Bunnenberg, E., Linder. R.
E., LaMar, G. N.. Del Gaudio, J., Lang, G., Spartalian, K.: J. Am. Chem. Soe 102, 4182
(1980)
76. Mashiko, T., Reed, C A., Haller, K. J., Kastner, M. E., Scheidt, W. R.: ibid. 103, 5758
(1981)
77. Walker, F. A.: ibid. 102, 3254 (1980)
78. Walker, F. A., Benson, M.: ibid. 102, 5530 (1980)
79. Santon, R. J., Wilson, L. J.: J. Chem. Soe, Chem. Commun., 359 (1984)
80. Molinaro, F. S., Little, R. G., bers, J. A.: J. Am. Chem. Soe 99, 5628 (1977)
81. Goff, H.: ibid. 102, 3252 (1980)
82. Traylor, T. G., Mincey, T. C Berzinis, A. P.: ibid. 103, 7084 (1981)
83. Collman, J. P., Groh, S. E.: ibid. 104, 1391 (1982)
84. Buckingham, D. A., Rauchfuss, T. B.: J. Chem. Soe, Chem. Commun., 705 (1978)
201
86. Smith K. M., Bisset, G. M. F.: J. Chem. Soc., Perkin Trans. 1, 2625 (1981)
87. Kong, J. L. Y., Loach, P. A.: J. Heterocycl. Chem. 17, 737 (1980)
88. Mcintosh, A. R., Siemiarczuk, A., Bolton, J. R., Stillman. M. J., Ho, T.-F., Weedon, A. C:
J. Am. Chem. Soc 105, 7215 (1983)
89. Mcintosh, A. R., Siemiarczuk, A., Bolton, J. R., Stillman, M. J., Ho, T.-F.. Weedon, A. C:
ibid. 105, 7224 (1983)
90. Wang, C-B., Tien, J. T., Lopez, J. R., Lui, Q.-Y., Joshi, N. B., Hu, Q.-Y.: Photobiochem.
Photobiophys. 4, 177 (1982)
91. Joshi, N. B., Lopez, J. R., Tien, H. T., Wang. C-B., Lui, Q.-Y.: J. Photochem. 20, 139
(1982)
92. Tabushi. I., Koga, N.. Yanagita, M.: Tetrahedron Lett., 257 (1979)
93. Wasielewski, M. R., Niemczyk, M. P.: J. Am. Chem. Soc 106, 5043 (1984)
94. Wasielewski, M. R., Niemczyk, M. P., Svec, W. A., Pewitt, E. B.: ibid. 107, 1080 (1985)
95. Joran, A. D., Leland, B. A.. Geller, G. G., Hopfield, J. J., Dervan, P. B.: ibid. 106, 6090
(1984)
96. Nishitani, S., Kurata, N., Sakata, Y.. Misumi, S., Migita, M., Mataga, N.: Tetrahedron Lett.
22,2099 (1981)
97. Mataga, N., Karen, A., Okada, T., Nishitani, S., Kurata, N., Sakata, Y., Misumi, S.: J. Phys.
Chem. 22, 5138 (1984)
98. Nishitani, S., Kurata, N., Sakata, Y., Misumi, S., Karen. A., Okada, T., Mataga, N.: J. Am.
Chem. Soc 105, (1983)
99. Moore, T. A., Gust, D., Mathis, P., Mialocq, J.-C, Chachaty, C, Bensasson. R. V., Land,
E. J., Doizi, D., Liddel, P. A., Lehman, W. R., Neme h, G. A., Moore, A. L.: Nature
(London) 307, 630 (1984)
100. Maiya, G. B.. Krishnan, V.: Inorg. Chim. Acta 77, L13 (1983)
101. Kobayashi, N., Akiba, V.. Takatori, K., Veno, A., Osa, T.: Heterocycles 19, 2011 (1982)
100. Eshima, K., Matsushita, Y., Sekine. M., Nishide, H., Tsuchida, E.: Nippon Kayaku Kaishi.
214 (1983)
101. Lown, J. W., Joshua, A. V.: J. Chem. Soc., Chem. Commun., 1298 (1983)
102. Hashimoto, Y., Lee, C-S., Shudo, K., Okamoto, T.: Tetrahedron Lett. 24, 1523 (1983)
103. Collman, J. P., Gagne, R. R., Halbert, T. R., Marchon, J.-C, Reed, C A.: J. Am. Chem.
Soc 95, 7868 (1973)
104. Collman, J. P., Gagne, R. R., Reed, C A., Halbert, T. R., Lang, G., Robinson, W. T.: ibid.
96, 1427 (1975)
105. Anzui, K., Hatano, K.: Chem. Pharm. Bull. 32, 1273 (1984)
106. Freilag, R. A., Whitten, D. G.: J. Phys. Chem. 87, 3918 (1983)
107. Freilag, R. A., Whitten, D. G.: J. Am. Chem. Soe 103, 1226 (1981)
108. Collman, J. P., Gagne, R. R., Reed, C A., Robinson, W. T., Rodley, G. A.: Proe Natl.
Acad. Sci. U.S.A. 71, 1326 (1974)
109. Jameson, G. B., Molinaro. F. S., Ibers, J. A., Collman, J. P., Brauman, J. I., Rose, E.,
Suslick, K. S.: J. Am. Chem. Soe 102, 3224 (1980)
110. Collman, J. P., Gagne, R. R., Gray, H. B., Hare, J. W.: ibid. 96, 6522 (1974)
111. Collman, J. P., Brauman, J. I., Halbert, T. R., Suslick, K. S.: Proe Natl. Acad. Sci. U.S.A.
73, 3333 (1976)
112. Gmai, H., Nakata, K., Nakatsubo, A., Kakagawa, S.. Vcrmori, Y., Kyuno, E.: Synth. React.
Inorg. Met.-Org. Chem. 13, 761 (1983)
113. Baldwin, J. E., Perlmutter, P.: Top. Curr. Chem. 121 (Host Guest Complex Chem. 3), 181
(1984)
114. David, S., Dolphin, D., James. B. R.: in Frontiers of Bioinorganic Chemistry (ed. Xavier, A.
V.), VCH Verlagsgesellschaft, Weinheim, pp. 163-182 (1985)
115. Collman, J. P., Gagne. R. R.. Reed, C A.: J. Am. Chem. Soe 96, 2629 (1974)
116. Collman, J. P., Brauman, J. I., Suslick, K. S.: ibid. 97, 7185 (1975)
117. Bogatskii, A. V., Zhilina, Z. I., Danilina, N. I.: Dokl. Akad. Nauk. S.S.S.R. (Engl. Ed.)252,
127 (1980)
118. Bogatskii, A. V., Zhilina, Z. I., Krasnoshchekaya, S. P.. Zakharova, R. M.: Zh. Org. Khim.
Engl. Ed. 18, 2035 (1982)
119. Valiotti, A., Adeyemo, A., Williams, F. A., Ricks, L., North, J., Hambright, P.: J. Inorg.
Nucl. Chem. 43, 2653 (1981)
202
121. Buckingham, D. H., Clark, C R., Webley, W. S.: J. Chem. Soc Chem. Commun., 192
(1981)
122. Buckingham, D. A., Gunter, M. G.. Mander, L. N.: J. Am. Chem. Soc 100, 2899 (1978)
123. Gunter, M. G., Mander, L. N., McLaughlin, G. M., Murray, K. S.. Berry, K. J., Clark, P. E.,
Buckingham, D. A.: ibid. 102, 1470 (1980)
124. Elliott. C M.. Krebs, R. R.: ibid. 104, 4301 (1982)
125. Groves, J. T., Myers, R. S.: ibid. 105, 5791 (1983)
126. Tabushi. I., Kodera. M.. Yokoyama, M.: ibid. 107, 4466 (1985)
127. Lecas-Nawrocka. A., Levisalles, J., Mariacher, C, Renko, Z., Rose, E.: Can. J. Chem. 62,
2054 (1984)
128. Collman, J. P., Brauman. J. I., Doxsee, K. M.. Sessler, J. L.. Morris. R. M.. Gibson, O. H.:
Inorg. Chem. 22, 1427 (1983)
129. Collman, J. P.. Brauman, J. I., Doxsee, K. M., Halbert, T. R.,Suslick, K. S.: Proc. Natl.
Acad. Sci. U.S.A. 75, 564 (1978)
130. Collman, J. P., Brauman, J. I., Doxsee. K. M.: ibid. 76, 6035 (1979)
131. Almog, J., Baldwin, J. E., Dyer, R. L., Peters, M.: J. Am. Chem. Soc 97, 226 (1975)
132. Almog, J.. Baldwin, J. E., Crossley, MK. J., De Bernardis, J. F., Dyer, R. L., Huff, J. R.,
Peters, M. K.: Tetrahedron 37, 3589 (1981)
133. Ellis, Jr., P. E., Linard, J. E., Szymanski, T., Jones. R. D., Budge, J. R., Basolo, F.: J. Am.
Chem. Soc 102, 1889 (1980)
134. Budge, J. R., Ellis, Jr., P. E.Jones, R. D., Linard, J. E., Basolo, F., Baldwin. J. E., Dyer. R.
L.: ibid. 101, 4760 (1979)
135. Ellis, Jr., P. E., Linard, J. E., Jones, R. D., Budge, J. R., Basolo, F.: ibid. 102, 1896 (1980)
136. Hashimoto, T., Dyer, R. L., Crossley, M. J., Baldwin. J. E., Basolo, F.: ibid. 104, 2101 (1982)
137. Baldwin, J. E., Crossley, M. G., De Bernardis, J.: Tetrahedron 38, 685 (1982)
138. Shimizu, M., Basolo, F., Vallejo, M. N., Baldwin, J. E.: Inorg. Chim. Acta 91, 247 (1984)
139. Shimizu, M., Basolo, F.. Vallejo, N. M., Baldwin. J. E.: ibid. 91, 251 (1984)
140. Rose, E. J., Vankatasurbramanian, P. N., Swartz, J. C, Jones, R. D., Basolo, F., Hoffman,
B. M.: Proc. Natl. Acad. Sci. U.S.A. 79, 5742 (1982)
141. Jameson, G. B., Ibers, J. A.: J. Am. Chem. Soc 102, 2823 (1980)
142. Sabat, M., Ibers, J. A.: ibid. 104, 3715 (1982)
143. Clayden, N. J., Moore, G. R., Williams, R. J. P., Baldwin. J. E., Crossley, M. J.: J. Chem.
Soe, Perkin Trans. 2, 1693 (1982)
144. Clayden, N. J., Moore, G. R.. Williams. R. J. P.. Baldwin. J. E., Crossley, M. J.: ibid. 2, 1863
(1983)
145. Baldwin, J. E., Cameron, J. H., Crossley, M. J., Dayley, E. J.: J. Chem. Soe, Dalton Trans.,
1739 (1984)
146. Collman, J. P., Brauman, J. I., Collins, T. J., Iverson, B. L., Sessler, J. L.: J. Am. Chem. Soe
103, 2450 (1981)
147. Collman, J. P., Brauman, J. I., Collins, T. J.. Iverson, B. L., Lang. G., Pettman. R. B.,
Sessler, J. L., Walters. M. A.: ibid. 105, 3038 (1983)
148. Collman, J. P., Brauman. J. I., Iverson, B. L., Sessler, J. L., Morris, R. M.. Gibson, O. H.:
ibid. 105, 3052 (1983)
149. Amundsen, A. R., Vaska, L.: Inorg. Chim. Acta. 14, L49 (1975)
150. Suslick, K. S., Fox, M. M.: J. Am. Chem. Soe 105, 3507 (1983)
151. Lindsey, J. S., Mauzerall, D. C: ibid. 104, 4498 (1982)
152. Lindsey, J. S., Mauzerall. D. C, Linschitz, H.: ibid. 105, 6528 (1983)
153. Ogoshi, H., Sugimoto, H.. Yoshida, Z.: Tetrahedron Lett., 4481 (1976)
154. Ogoshi, H., Sugimoto, H.. Yoshida, Z.: ibid. 1515 (1977)
155. Ogoshi, H., Sugimoto, H., Miyake, M., Yoshida, Z. I.: Tetrahedron 40, 579 (1984)
156. Battersby, A. R., Buckley, D. G., Hartley, S. G., Turnbull, M.: J. Chem. Soe, Chem.
Commun., 879 (1976)
157. Chang, C. K., Kuo, M.-S.: J. Am. Chem. Soe 101, 3413 (1979)
158. Ward, B., Wang, C-B., Chang, C K.: ibid. 103, 5236 (1981)
159. Yu. N.-T., Kerr, E. A., Ward, B., Chang, C K.: Biochemistry 22, 4534 (1983)
160. Baldwin, J. E.. Klose, T., Peters. M.: J. Chem. Soe, Chem. Commun.. 881 (1976)
161. Baldwin, J. E., Crossley, M. J., Klose. T.. O'Rcar III, E. A., Peters, M. K.: Tetrahedron^,
27 (1981)
203
163. Wijesekera. T. P.: Ph.D. Thesis. University of British Columbia 1980
164. Wijesekera. T. P., Paine III, J. B., Dolphin. D.. Einstein, F. W. B.. Jones, T.: J. Am. Chem.
Soc. 105, 6747 (1983)
165. Diekmann, H.. Chang, C K.. Traylor, T. G.: ibid. 93, 4068 (1971)
166. Traylor, T. G.. Campbell. D.. Tsuchiya, S.: ibid. 101, 4748 (1979)
167. Traylor, T. G.. Campbell, D., Tsuchiya, S., Mitchell, M., Stynes, D. V.: ibid. 102, 5939 (1980)
168. Travlor. T. G.. Mitchell. M. J . Tsuchiya. S., Campbell. D. H.. Stynes, D. V., Koga. N.: ibid.
103. 5234 (1981)
169. Travlor. T. G.. Tsuchiva. S.. Campbell. D.. Mitchell. M., Stvncs, D.. Koga. N.: ibid. 107, 604
(1985)
170. Chanii. C K.. DiNeIIo. R. K.. Dolphin. D.: in Inorganic Syntheses (ed. Busch. D. H.). John
Wilev & Sons. New York. Vol. XX. 147 (1980)
171. David. S.. Dolphin, D., James. B. R., Paine. J. B. Ill, Wijesekera. T. P.. Einstein. F. W. B..
Jones. T.: Can. J. Chem. 64. 208 (1986)
172. David. S.: Ph.D. Thesis. University of British Columbia 1985
173. Travlor. T. G.. Koga. N.. Dearduff. L. A.. Swepston, P. N., bers. J. A.: J. Am. Chem. Soc
106, 5132 (1984)
174. Travlor. T. G.. Koga. N.. Dearduff. L. A.: ibid. 107. 6504 (1985)
175. Morgan. B.: Ph.D. Thesis. University of British Columbia 1984
176. Morgan, B., Dolphin, D.: Angew. Chem. Int. Ed. 24, 1003 (1985)
177. Battersby, A. R., Hartley. S. G., Turnbull, M. D.: Tetrahedron Lett., 3169 (1978)
178. Cruse, W. B., Kennard. O., Sheldrick, G. M., Hamilton. A. D., Hartley. S. G., Battersby, A.
R.: J. Chem. Soc, Chem. Commun., 700 (1980)
179. Battersby, A. R., Howson, W., Hamilton, A. D.: ibid. 1266 (1982)
180. Chang. C K.: J. Am. Chem. Soc. 99, 2819 (1977)
181. Richardson, N. M., Sutherland, I. O.. Camilleri. P., Page, J. A.: Ictrahedron Lett. 26, 3739
(1985)
182. Bogatskii, A. V.. Zhilina, Z. I., Stepanov, D. E.: Zh. Org. Khim. Engl. Ed. 18, 2039 (1982)
183. Hamilton, A. D., Lehn, J.-M., Sessler, J. L.: J. Chem. Soc., Chem. Commun., 311 (1984)
184. Chang. C K., Koo, M. S., Ward, B.: ibid. 716 (1982)
185. Guntcr. M. J.. Mander, L. N.: J. Org. Chem. 46, 4792 (1981)
186. Gunter, M. J., Mander, L. N., Murray. K. S., Clark, P. E.: J. Am. Chem. Soc. 103, 6784
(1981)
187. Ganesh, K. N., Sanders, J. K. N.: J. Chem. Soc, Chem. Commun., 1129 (1980)
188. Ganesh, K. N.. Sanders, J. K. N.: J. Chem. Soc, Perkin Trans. 1, 1611 (1982)
189. Ganesh. K. N.. Sanders. J. K. N.. Waterton. J. C: ibid. 1. 1617 (1982)
190. Sanders. J. K., Leighton, P.: J. Chem. Soc, Chem. Commun., 24 (1985)
191. Leighton, P., Sanders, J. K. M.: ibid. 854 (1984)
192. Abraham. R. J., Leighton, P., Sanders. J. K. M.: J. Am. Chem. Soc 107, 3472 (1985)
193. Leighton, P.. Sanders. J. K. M.: J. Chem. Soc. Chem. Commun.. 856 (1984)
194. Hamilton. A. D.. Rubin, H.-D.. Bocarsly. A. B.: J. Am. Chem. Soc 106, 7255 (1984)
195. Momenteau. M.. Mispclter. J., Loock. B.. Bisagni. E.: J. Chem. Soc. Perkin Trans. 1, 189
(1983)
196. Momenteau. M.. Loock, B.. Mispelter, J.. Bisagni, E.: Nouv. J. Chem. 3, 77 (1979)
197. Lexa, D., Momcnteau. M., Rentien, P., Rytz, G., Saveant, J. M.. Xu. F.: J. Am. Chem. Soc
106, 4755 (1984)
198. Becker. J. Y.. Dolphin, D., Paine. J. B. III. Wijeskera, T. P.: J. Electroanal. Chem. 164, 335
(1984)
199. Zhilina. Z. I., Bogatskii, A. V.. Vodzinskii, S. V., Abramovich, A. E.: Zh. Org. Khim. Eng.
Ed. 18, 227 \ (1982)
200. Wciser, J., Staab, H. A.: Angew. Chem. Int. Ed. 23, 623 (1984)
201. Momenteau, M.. Lavalette D.: J. Chem. Soc, Chem. Commun., 341 (1982)
202. Mispelter. J.. Momcnteau, M., Lavalette, D. Lhoste. J.-M.: J. Am. Chem. Soc 105, 5165
(1983)
203. Momcnteau, M., Loock, B.. Lavalette, D.. Tetreau, C1 Mispelter, J.: J. Chem. Soc, Chem.
Commun., 962 (1983)
204. Battersby, A. R., Hamilton, A. D.: ibid. 117 (1980)
205. Battersby, A. R., Bartholomew. S. A. J., Nitta. J.: ibid. 1291 (1983)