Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
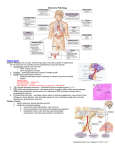
PATHOLOGY OF ENDOCRINE SYSTEM 2009 Dr. Huda M Zahawi, FRC.Path The Endocrine system is divided into : • Endocrine organs dedicated to production of hormones e.g. pituitary,thyroid….etc • Endocrine components in clusters in organs having mixed functions e.g. pancreas, ovary, testes….. • Diffuse endocrine system comprising scattered cells within organs acting locally on adjacent cells without entry into blood stream Disease divided into : 1- Diseases of overproduction of secretion ( Hyperfunction ) 2- Diseases of underproduction ( Hypofunction ) 3- Mass effects ( Tumors ) N.B. Correlation of clinical picture , hormonal assays , biochemical findings , together with pathological picture are of extreme importance in most conditions. PITUITARY GLAND PITUITARY GLAND • Pituitary in sella turcica,& weighs about 0.5gm. • Connected to the HYPOTHALAMUS with stalk. • Composed of : A-ADENOHYPOPHYSIS- (80%) – – Blood supply is through portal venous plexus Hypothalamic-Hypophyseal feed back control B- NEUROHYPOPHYSIS – – – From floor of third ventricle Modified glial cells & axons hypothalamus. Has its own blood supply. CELLS & SECRETIONS : A- Anterior pituitary ( Adenohypophysis ) 1-Somatotrophs from acidophilic cells → Growth H. 2- Lactotrophs from chromophobe cells → Prolactin 3- Corticotrophs from basophilic cells → ACTH,MSH . 4- Thyrotrophs from pale basophilic cells → TSH 5- Gonadotrophs from basophilic cells → FSH, LH B- Posterior pituitary ( Neurohypophysis ) 1- Oxytocin 2- ADH HYPERPITUITARISM & PITUITARY ADENOMA In most cases, excess is due to ADENOMA arising in the anterior lobe. Less common causes include : * Hyperplasia * Carcinoma * Ectopic hormone production * Some hypothalamic disorders Pathogenesis of pituitary adenomas : • Mutations in G-proteins ( α subunit) in the GNAS1 gene on chromosome 20q13 lead to activation • 40% of GH secreting adenomas & less in ACTH • G-proteins involved in signal transduction : GDP G proteins GTP cAMP GTPase • Mutations in α subunit interfere with GTPase function • Mutations in RAS, overexpression in C- MYC & NM23 inactivation found in more aggressive tumor • Other mutations : MEN-1 gene ( Menin) Features common to all pituitary adenomas : • 10% of all intracranial neoplasms & 25% incidental 3% occur with MEN syndrome • 30-50 years of age • Primary pituitary adenomas usually benign • May or may not be functional • If functional, the clinical effects are secondary to the hormone produced. • More than one hormone may be produced by same cell • Although most are localized, invasive adenomas erode sella turcica & extend into cavernous & sphenoid sinus CLINICAL FEATURES of PITUITARY ADENOMA: 1- Symptoms of hormone produced 2- Local mass effects : i- Radiological changes ii-Visual field abnormalities iii-Elevated intracranial pressure 3- Hypopituitarism 4- Pituitary apoplexy Mass effect of pituitary adenoma Downloaded from: Robbins & Cotran Pathologic Basis of Disease (on 4 December 2005 01:50 PM) Morphology of pituitary adenomas : • Well circumscribed,invasive in up to 30% • Size 1cm. or more, specially in nonfunctioning tumor • Hemorrhage & necrosis seen in large tumors Microscopic picture : • Uniform cells, one cell type (monomorphism) • Absent reticulin network • Rare or absent mitosis Sella turcica with pituitary adenoma Uniform cells of pituitary adenoma Downloaded from: Robbins & Cotran Pathologic Basis of Disease (on 4 December 2005 01:50 PM) Types of Pituitary Adenomas • Previously classified according to histological picture e.g : Acidophilic Adenoma • Now according to immunohistochemical findings & clinical picture ….. e.g. Growth hormone secreting adenoma Immunoperoxidase for GH 1- PROLACTINOMA : • 30% of all adenomas, chromophobe or weakly acidophilic • Functional even if small, but related to size • Other causes of prolactin include : estrogen therapy, pregnancy, reserpine , hypothyroidism…… • Any mass in the suprasellar region may interfere with normal prolactin inhibition Prolactin ( STALK EFFECT ) • Mild elevation of prolactin does NOT always indicate prolactin secreting adenoma ! Symptoms : • • • • Galactorrhea Amenorrhea Decrease libido Infertility 2- Growth hormone secreting adenoma : • 40% Associated with GNAS 1 gene mutation • Persistent secretion of growth hormone leads to secretion of Insulin – like GF → symptoms • Composed of granular ACIDOPHILIC cells • May be mixed with prolactin secretion. • Symptoms delayed so adenomas are usually large • Produce GIGANTISM or ACROMEGALLY • Other symptoms : diabetes, arthritis, large jaw & hands, osteo porosis, BP, HF…..etc 3- Corticotroph cell adenoma • • • • • Usually microadenomas Higher chance of becoming malignant Chromophobe or basophilic cells Functionless or Cushing ‘s Disease ( ACTH ) Bilateral adrenalectomy or destruction may result in aggressive adenoma: Nelson’s Syndrome • Corticotroph microadenoma Macroadenoma • ICP 4- Non functioning adenoma 20% silent or null cell ,nonfunctioning & produce mass effect only 5- Gonadotroph producing LH &FSH- ( 10-15%)Function silent or is minimal , late presentation mainly mass effect produced. Produce gonadotrophin α subunit, β- FSH & β-LH 6- TSH producing ,(1%) rare cause of hyperthyroidism 7- Pituitary carcinoma - Extremely rare, diagnosed only by metastases. HYPOPITUITARISM : • Loss of 75% of ant. Pituitary Symptoms • Congenital or acquired, intrinsic or extrinsic • Symptoms include dwarfism, & effect of individual hormone deficiencies. Loss of MSH → Decreased pigmentation • Acquired causes include : 1- Nonsecretory pituitary adenoma 2- Ischemic necrosis e.g. SHEEHAN’S SYNDROME (post partum hmg.) sickle cell anemia, DIC, Pituitary apoplexy… 3- Iatrogenic by radiation or surgery 4- Autoimmune ( lymphocytic) hypophysitis 5- Inflammatory e.g sarcoidosis or TB ….. 6- Empty Sella Syndrome : Radiological term for enlarged sella tursica, with atrophied or compressed pituitary. May be primary due to downward bulge of arachnoid into sella floor compressing pituitary. Secondary is usually surgical. 7- Infiltrating diseases in adjacent bone e.g. Hand Schuller – Christian Disease 8- Craniopharyngioma Craniopharyngioma : * 1-5 % of intracranial neoplasms * Derived from remnants of Rathke’s Pouch * Suprasellar or intrasellar ,often cystic with calcification * Children or adolescents most affected * Symptoms may be delayed ≥ 20yrs( 50%) * Symptoms of hypofunction or hyperfunction of pituitary and /or visual disturbances, diabetes insipidus * Benign & slow growing POSTERIOR PITUITARY SYNDROMES: 1-A- ADH deficiency causes Diabetes Insipidus Excessive urination,dilute urine , due to inability to reabsorb water from the collecting tubules. Causes include head trauma, tumors & inflammations in pituitary or hypothalamus…etc. B- Syndrome of inappropriate ADH secretion Causes excessive resorption of water hyponatremia e.g Small Cell CA of Lung 2-Abnormal oxytocin secretion : Abnormalitis of synthesis & release have not been associated with any significant abnormality. THYROID GLAND • Development from evagination of pharyngeal tissue into neck • Abnormal descent Lingual thyroid , subhyoid, substernal • Weight 15-20gm. Responsive to stress • Structure : varying sized follicles lined by columnar epithelium , filled with colloid, interfollicular C cells • Secretion of T3 & T4 is controlled by trophic factors from hypothalamus & ant.pituitary THYROTOXICOSIS: • Hypermetabolic state caused by T4, T3. A- Associated with hyperthyroidism: Primary : Graves Disease Toxic multinodular goiter Toxic adenoma Secondary : TSH secreting pit. adenoma B- Not associated with hyperthyroidism : Thyroiditis Struma ovarii Exogenous thyroxine intake Clinical Picture related to Sympathetic Stimulation • Constitutional symptoms : heat intolerance, sweating, warm skin, appetite but ↓weight • Gastrointestinal : hypermotility, malabsorption • Cardiac : palpitation, tachycardia, CHF • Menstrual disturbances • Neuromuscular : Tremor, muscle weakness • Ocular : wide staring gaze, lid lag, thyroid ophthalmopathy • Thyroid storm : severe acute symptoms of sympathetic overstimulation • Apathetic hyperthyroidism : incidental Diagnosis of Hyperthyroidism : • Measurement of serum TSH (↓ ) + free T4 is the most useful screening test for thyrotoxicosis • TSH level is normal or in secondary thyrotoxicosis • In some patients , T3 but T4 normal or ↓ • Measurement of Radioactive Iodine uptake is a direct indication of activity inside the gland Normal radioactive I uptake HYPOTHYROIDISM : Primary : 1- Loss of thyroid tissue due to surgery or radiation Rx. 2- Hashimoto’s thyroiditis 3- Iodine deficiency specially in endemic areas 4- Primary idiopathic hypothyroidism 5- Congenital enzyme deficiencies 6- Drugs e.g. iodides, lithium….. 7- Thyroid dysgenesis ( developmental ) Secondary : Pituitary or hypothalamic failure Hypothyroidism is commoner in endemic areas of iodine deficiency CRETINISM : hypothyroidism in infancy & is related to the onset of deficiency . If early in fetal life Mental retardation , short stature, hernia, skeletal abnormalities, MYXEDEMA in adults Apathy, slow mental processes, cold intolerence,accumulation of mucopolysaccharides in subcutaneous tissue Lab.tests : TSH in primary hypothyroidism, unaffected in others T4 in both. THYROIDITIS : • Mostly autoimmune mechanisms • Microbial infection is rare • Types include : 1- Chronic lymphocytic ( Hashimoto’s ) thyroiditis 2- Subacute granulomatous ( de Quervain) thyroiditis 3- Subacute lymphocytic thyroiditis 4- Riedel thyroiditis 5- Palpation thyroiditis HASHIMOTO’s THYROIDITIS : Chronic Lymphocytic Thyroiditis • Autoimmune disease characterized by progressive destruction of thyroid tissue • Commonest type of thyroiditis • Commonest cause of hypothyroidism in areas of sufficient iodine levels • F:M = 10-20 :1, 45-65 yrs. • Can occur in children Pathogenesis : A - T cell sensitization to thyroid antigens 1- Sensitized CD4 T cells Cytokine mediated ( IFN- γ)cell death inflammation,macrophage activation 2- CD8+ cytotoxic T cell mediated cell death: Recognition of AG on cell killed 3- Presence of thyroid AB Antibody dependent cell mediated cytotoxicity by NK cells B- Genetic predisposition : ↑ in relatives of 1st.degree Association with HLA – DR 3 & DR- 5 Morphology: • Gland is a smooth pale goitre, minimally nodular, well demarcated. • Microscopically : - Dense infiltration by lymphocytes & plasma cells - Formation of lymphoid follicles, with germinal centers - Presence of HURTHLE CELLS - With or without fibrosis • Clinically : – Painless symmetrical diffuse goiter – May show initial toxicosis ( Hashitoxicosis ). – Later marked hypothyroidism. – Patients have risk of B-Cell lymphoma Downloaded from: Robbins & Cotran Pathologic Basis of Disease (on 4 December 2005 01:50 PM) © 2005 Elsevier SUBACUTE GRANULOMATOUS THYROIDITIS : • Middle aged , more in females. Viral etiology ? • Self-limited (6-8w) • Acute onset of pain in the neck , fever, ESR, WBC • Transient thyrotoxicosis. • Morphology : – Firm gland. – Destruction of acini leads to mixed inflammatory infiltrate. – Neutrophils , Macrophages & Giant cells & formation of granulomas SUBACUTE LYMPHOCYTIC THYROIDITIS : (Silent) • Middle aged females & post partum patients • Probably autoimmune with circulating AB • May recur in subsequent pregnancies • May progress to hypothyroidism • Histology similar to Hashimoto’s thyroiditis without Hurthle cell metaplasia • Reidel’s Thyroiditis – Dense fibrosis without prominent inflammation ? Considered as fibromatosis rather than thyroiditis GRAVE’S DISEASE : • Commonest cause of endogenous hyperthyroidism • Age 20- 40 yrs., • M: F ratio is 1: 7 • More common in western races Main features of GRAVES DISEASE : 1 - Thyrotoxicosis with smooth symmetrical enlargement of thyroid 2 - Infiltrative ophthalmopathy with exophthalmus in 40% 3- Pretibial myxedema in a minority • Lab findings : T4, T3 , TSH • Radioactive study: Diffuse uptake of radioactive I Pathogenesis of GRAVE’S DISEASE : • • • • • Genetic etiology + Autoimmune processes GENETIC EVIDENCE : May be familial 60% concordance in identical twins Susceptibility is associated with HLA-B8 & - DR3 • May exist with other similar diseases e.g. SLE, Pernicious anemia, Diabetes type I, Addison’s dis. IMMUNE MECHANISMS : • Antibodies to thyroid peroxisomes & thyroglobulin • Patients develop autoantibodies to TSH receptor – Thyroid Stimulating Immunoglobulin ( TSI) binds to TSH receptor → thyroxin *** – Thyroid Growth Stimulating Immunoglobulin (TGI) → proliferation of thyroid epithelium – TSH binding inhibitor immunoglobulins (TBIIs) prevent TSH from binding to receptor • Both stimulation & inhibition may coexist Morphology : • Smooth enlargement of gland with diffuse hyperplasia & hypertrophy • Lining epithelium of acini : Tall & hyperplastic ± papillae • Colloid : Minimal thin colloid with scalloped edge Downloaded from: Robbins & Cotran Pathologic Basis of Disease (on 4 December 2005 01:50 PM) © 2005 Elsevier Changes in Extrathyroid tissue : • Generalized lymphoid hyperplasia • Ophthalmopathy : Edematous orbital muscles &infiltration by lymphocytes followed by fibrosis • Thickening of skin & subcutaneous tissue • Accumulation of glycosaminoglycans which are hydrophilic • Result : Displacement of eyeball & exophthalmus → redness, dryness, ulceration, infection in conjunctiva • Cause : Expression of aberrant TSH receptor responding to circulating anti TSH receptor AB → inflammatory lymphocytic reaction Downloaded from: Robbins & Cotran Pathologic Basis of Disease (on 15 December 2005 07:30 AM) © 2005 Elsevier DIFFUSE NONTOXIC & MULTINODULAR GOITRE GOITER = Enlargement of thyroid Most common cause is iodine deficiency impaired hormone synthesis TSH hypertrophy & hyperplasia of follicles Goiter Endemic : 10% of population have goiter Sporadic : 1- Physiological demand 2- Dietary intake of excessive calcium & cabbages…etc 3- Hereditary enzyme defects MORPHOLOGY : • Initially diffuse → nodular with degenerative changes: colloid cysts, hemorrhage, fibrosis, calcification • If large may extend retrosternally • Pressure symptoms are a common complaint • Picture is that of varying sized follicles, hemorrhage , fibrosis , cysts, calcification • Patient is often EUTHYROID. but may be toxic or hypofunctioning. Normal radioactive I uptake Downloaded from: Robbins & Cotran Pathologic Basis of Disease (on 4 December 2005 01:50 PM) © 2005 Elsevier NODULES in the thyroid : • Nodules in thyroid may be multiple or solitary • Any solitary nodule in the thyroid has to be investigated as some are neoplastic. Investigations include FNA , Radioactive image technique, Ultrasound, & (T4,T3 & TSH ) levels • HOT nodule takes up radioactive substance ( functional) • COLD nodule does not it take up ( nonfunctional ) Downloaded from: Robbins & Cotran Pathologic Basis of Disease (on 4 December 2005 01:50 PM) © 2005 Elsevier General rules of nodules in the thyroid : 1- Solitary nodule is MORE likely to be NEOPLASTIC than multiple 2- Hot nodules are more likely to be BENIGN 3- Not every cold nodule is malignant . Many are nonfuctioning adenomas, or colloid cysts , nodules of nodular goitre….etc Up to 10% of cold nodules prove to be malignant. 4- Nodules in younger patients are more likely to be NEOPLASTIC 5- Nodules in males are more likely to be NEOPLASTIC . 6- History of previous radiation to the neck is associate with increased risk of malignancy NEOPLASMS of the THYROID : ADENOMAS: • Usually single. • Well defined capsule • Commonest is follicular± Hurthle cell change • May be toxic • Size 1- 10cm. Variable colour • Activating somatic mutation in TSH receptor is identified leading to overproduction of cAMP • 20% have point mutation in RAS oncogene Microscopical Picture : • 1- Uniform follicles , lined by cuboidal epithelial cells. • 2- Focal nuclear pleomorphism, nucleoli …. ( Endocrine atypia ) • 3- Presence of a capsule with tumor compressing surrounding normal thyroid outside . * Integrity of capsule is important in differentiating adenoma from well differentiated follicular carcinoma. • Capsular and/ or vascular invasion →Carcinoma Downloaded from: Robbins & Cotran Pathologic Basis of Disease (on 4 December 2005 01:50 PM) © 2005 Elsevier Adenoma with intact capsule © 2005 Elsevier Capsular invasion) CARCINOMAS of THYROID : • Incidence about 1-2% of all malignancies. • Wide age range ,depending on type. • Generally commoner in females, but in tumors occurring in children or elderly , equal incidence in both sexes. • Most are derived from follicular cells • Few are derived from ‘C’ cells TYPES of THYROID CARCINOMA : 1- Papillary Carcinoma ( 75- 85% ),any age,but usual type in children. 2- Follicular Carcinoma ( 10- 20% )More in middle age 3- Medullary Carcinoma ( 5% ) age 50-60 but younger in familial cases with MEN syndrome 4- Anaplastic Carcinoma ( 5% ) , old age Presenting symptom is usually a mass , maybe incidental in a multinodular goitre specially papillary, & follicular Pathogenesis of Thyroid Cancer : 1- Genetic lesions : Most tumors are sporadic Familial is mostly Medullary CA , Papillary CA • Papillary CA : – Chromosomal rearrangement in tyrosin kinase receptor gene (RET) on chr.10q11 ret/PTC tyrosine kinase activity ( 1/5 of cases specially in children) – Point mutation in BRAF oncogene (1/3-1/2) • Follicular Carcinoma : – RAS mutation in ½ of cases OR – PAX8- PPAR γ 1 fusion gene in 1/3 of cases • Medullary Carcinoma : – RET mutation Receptor activation • Anaplastic Carcinoma : – Probably arising from dedifferentiation of follicular or papillary CA inactivation of P53 2- Environmental Factors : • Ionizing radiation specially in first two decades • Most common is Papillary CA. with RET gene rearrangement 3- Preexisting thyroid disease : • Incidence of thyroid CA is more in endemic areas • Long standing multinodular goiter → Follicular CA • Hashimotos thyroiditis → Papillary CA & B cell lymphoma • TYPES OF THYROID CARCINOMAS PAPILLARY CARCINOMA : • Cold on Scan by radioactive Iodine • Solitary or multifocal • Solid or cystic, calcification • Composed of papillary architecture • Less commonly ‘Follicular Variant’ Downloaded from: Robbins & Cotran Pathologic Basis of Disease (on 4 December 2005 01:50 PM) © 2005 Elsevier Diagnosis based on NUCLEAR FEATURES • Nuclei are clear (empty) ,with grooves & inclusions ( Orphan Annie nuclei) • Psammoma bodies • Metastases mainly by L.N., sometimes from occult tumor • Hematogenous spread late & prognosis is GOOD FNA of Papillary CA (nuclear changes) Downloaded from: Robbins & Cotran Pathologic Basis of Disease (on 4 December 2005 01:50 PM) © 2005 Elsevier Psammoma body in Papillary CA FOLLICULAR CARCINOMA : • Usually cold but rarely functional ( warm ) • Well circumscribed with thick capsule (minimally invasive) or diffusely infiltrative • Composed of follicles , sometimes of Hurthle Cells • Diagnosis is based on CAPSULAR & VASCULAR invasion • Metastasize usually by blood Lungs, Bone, Liver ..etc. • Treatment by surgery Radioactive Iodine Thyroxin • Prognosis is not as good as papillary except in minimally invasive very well differentiated forms Follicular Carcinoma Downloaded from: Robbins & Cotran Pathologic Basis of Disease (on 4 December 2005 01:50 PM) © 2005 Elsevier Capsular invasion) Downloaded from: Robbins & Cotran Pathologic Basis of Disease (on 4 December 2005 01:50 PM) © 2005 Elsevier MEDULLARY CARCINOMA: • Arise from C cells CALCITONIN, CEA, serotonin, VIP • 80% Sporadic , or familial MEN Syndrome • Composed of polygonal or spindle cells , usually with demonstrable AMYLOID in the stroma • Calcitonin demonstrated in tumor cells • Level of calcitonin in serum may be useful for follow up • Family members may show C cell hyperplasia ,↑ Calcitonin, & RET mutation ( Marker for early diagnosis) • Metastases by blood stream • Prognosis intermediate , worse in MEN. 2B Medullary CA with amyloid Congo red for amyloid ANAPLASTIC CARCINOMA : • Elderly patients with multinodular goitre in 50% • Foci of papillary or follicular CA may be present in 20%- 30% , probable dedifferentiation process • Markedly infiltrative tumor , invading the neck → pressure on vital structures • Rapid progression, death within 1 year • Morphology : Composed of pleomorphic giant cells, spindle cells or small cell anaplastic varients, which may be confused with lymphoma • Radiosensitive tumor , no surgery • P53 mutation identified , consistent with tumor progression PARATHYROID GLAND • Derived from the third and fourth pharyngeal pouches. • 90% of people have four glands. • Location: mostly close to the upper or lower poles of the thyroid. • Can be found anywhere along the line of descent of the pharyngeal pouches. • There are two types of cells with intervening fat : - Chief & Oxyphil cells • Secretion of PTH is controlled by level of free calcium Hyperparathyroidism : Primary OR Secondary Primary Hyperparathyroidism: • Commonest cause of asymptomatic hypercalcemia • Female:Male ratio = 2-3 : 1. • Causes : Adenoma 75%-80% Hyperplasia 10-15% Carcinoma < 5% • Majority of adenomas are sporadic • 5% familial associated with MEN-1 or MEN-2A Genetic abnormalities : • PRAD 1 on chromosome 11 q cell cycle control cyclin D1 overexpression(10%-20%) • MEN 1 on 11q13 is a cancer suppressor gene - Germ line mutation in MEN-1 syndrome loss of function cell proliferation - *20% - 30% of sporadic cases may also show mutation of MEN1 *Either of above may cause tumor or hyperplasia • Biochemical findings : PTH , Ca , ↓ phosphate ,alkaline phosphatase • In other causes of hypercalcemia, PTH is ↓ Gland morphology in Hyperparathyroidism • Adenomas : • Usually single , rarely multiple • Well circumscribed, encapsulated nodule (0.5-5g.) • The cells are polygonal, uniform chief cells, few oxyphil cells. Adipose tissue is minimal in the tumor • Compressed surrounding parathyroid tissue in periphery, other glands normal or atrophic . • Hyperplasia : Enlargement of all 4 glands. Microscopically chief cell hyperplasia, or clear cell, usually, in a nodular or diffuse pattern. Note : Diagnosis of adenoma versus hyperplasia may depend on the size of the other glands Parathyroid carcinoma : • Larger than adenoma (5-10g) • Very adherent to surrounding tissue. • Pleomorphism & mitoses not reliable criteria for malignancy • Most reliable criteria for malignancy are : * Invasion **Metastases Morphology in other organs: • Skeletal system: – Bone resorption by osteoclasts, with fibrosis, cysts formation and hemorrhage Osteitis Fibrosa Cystica – Collections of osteoclasts form ‘ Brown Tumors” – Chondrocalcinosis and pseudogout may occur. • Renal system: – Ca. Stones. & Nephrocalcinosis. • Metastatic calcification in other organs: Blood vessels & myocardium , Stomach, Lung …etc Hyperparathyroidism, clinical picture • 50% of patients are asymptomatic. • Patients show Ca & PARATHORMONE levels in serum • Symptoms and signs of hypercalcemia: Musculoskeletal, Gastrointestinal tract, Urinary and CNS symptoms • Commonest cause of silent hypercalcemia . • In the majority of symptomatic hypercalcemia commonest cause is wide spread metastases to bone Painful Bones, Renal Stones, Abdominal Groans & Psychic Moans Downloaded from: Robbins & Cotran Pathologic Basis of Disease (on 4 December 2005 01:50 PM) © 2005 Elsevier Secondary Hyperparathyroidism : • Occur in any condition associated with chronic hypocalcemia, mostly chronic renal failure. • Glands are hyperplastic • Renal failure phosphate excretion increased serum phosphate, CaPTH Tertiary Hyperparathyroidism • Extreme activity of the parathyroid autonomous function & development of adenoma (needs surgery) Hypoparathyroidism : • Causes: – Damage to the gland or its vessels during thyroid surgery. – Idiopathic, autoimmune disease. – Pseudohypoparathyroidism, tissue resistance to PTH • Clinical features: -Tetany, convulsion, neuromuscular irritability, cardiac arrhythmias…… ENDOCRINE PANCREAS • Diseases mainly include : – Diabetes – Islet Cell Tumors DIABETES DIABETES : • Chronic disorder in which there is abnormal metabolism, of carbohydrate, fat & protein , characterized by either relative or absolute insulin deficiency, resulting in hyperglycemia. • Most important stimulus that triggers insulin synthesis from β cells is GLUCOSE • Other agents stimulate insulin release • Level of insulin is assessed by the level of C - peptide • Diagnosis : 1- Random glucose ≥ 200g / dL + symptoms 2- Fasting glucose of ≥ 126 / dL on more than one occasion 3- Abnormal OGTT when glucose level is more than 200g / dL 2hrs. after standard glucose load of 75 g. Classification : Causes could be Primary in the pancreas OR secondary to other disease conditions Primary diabetes is classified into : A- Type 1 B- Type 2 C- Genetic & Miscellaneous causes Whatever the type, complications are the same Type 1 :• Absolute deficiency of insulin due to β cell destruction ( 10%) • 90% of cells lost before metabolic changes appear • Age ≤ 20 yrs but may be latent • Normal or decreased weight • Ketoacidosis is common Type 2 : • Due to a combination of peripheral resistance to insulin action & inadequate secretory response by the pancreatic β cells • Commoner ( 80 - 90% ) • Insulin normal (relative insulin deficiency) • Patient is overweight • Rare ketoacidosis Type 3 : Miscellaneous causes • Genetic defects : – β cell function e.g. Maturity Onset Diabetes of the Young ( MODY)caused by a variety of mutations – Genetic defects of insulin processing or action e.g. Insulin gene or Insulin receptor mutations Secondary Miscellaneous Causes : • Diseases of exocrine pancreas e.g. chronic pancreatitis • Endocrinopathies e.g. Cushing’s Syndrome, Acromegally • Infections e.g. CMV • Drugs e.g. glucocorticoids • Gestational diabetes • Other genetic syndromes associated with diabetes PATHOGENESIS Pathogenesis of Type 1 Diabetes : 1- Genetic susceptibility 2- Autoimmunity 3- Environmental factors It is a combination of autoimmunity & environmental insult in a person with a known genetic susceptibility leading to destruction of β cells 1- Genetic susceptibility – Principal susceptibility genes located in region of MHC class II on chromosome 6p21 – 90% Associated with HLA- DR3,or HLADR4, or both – Racial predisposition, (Caucasians) but majority have no family history – 6- 20% familial ,< 40% in identical twins – Second susceptibility gene encodes a T cell inhibitory receptor (CTLA-4) interfering with normal T cell function 2- Autoimmunity - • Presence of CD 8+ & CD 4+ in islet cells “ Insulinitis” • Presence of islet cell antibodies ( insulin & GAD) in 80% of patients & in relatives several months or years before onset • Antibodies are highly selective against β cells • Relatives at risk have similar AB years before onset • 10% - 20% other autoimmune disease 3- Environmental factors An environmental insult may damage β cells rendering them antigenic. • • • Viruses : measles , coxsackie , rubella Chemicals Cow’s milk Pathogenesis of Type 2 diabetes : 1- Genetic factors 2- Insulin resistance & obesity 3 - cell secretion dysfunction 1- Genetic factors : – Genetic factors are more important than in type 1 diabetes, but this is multifactorial – 50% - 90% in identical twins – risk by 20%-40% in first degree relatives – No association with HLA & no autoimmune basis – Point mutation in insulin receptor identified affecting signaling pathway but rare ( 1-5%) 2 – Insulin resistance : • Decrease ability of peripheral tissue to respond to insulin Early : insulin resistance → insulin secretion due to compensatory of cell mass Later : relative insulin & cell mass to 20-50% MAIN FACTOR IN INSULIN RESISTANCE IS OBESITY Explanation : • Adipocytokines : – Resistin ↑ obesity → Insulin resistance – Leptin & Adiponectin contribute to insulin sensitivity but are ↓in obesity → resistance – PPAR γ is a nuclear receptor that regulates level of adipocytokines – FFA in tissues (lipotoxic effect) → insulin resistance 3- cell Dysfunction : • Defective glucose recognition due to ↑ intracellular levels of a mitochondrial protein ( UCP2) in β cells • Amylin : A protein normally produced by cells secreted with insulin in response to food ingestion Amylin accumulates outside cells, forming amyloid like deposits & may impair cell glucose sensing. Seen in up to 90% of cases of Type II diabetes Pathogenesis of complications : 1- Nonenzymatic glycosylation of proteins Glucose + Free amino acids Later → Irreversible combination Advanced Glycosylation End products =AGES Measured by level of glycosylated Hb ( HbA1c) AGES inactivate proteins & cross link with more proteins, deposited in vessels, renal glomeruli, …..etc Effects : Induce cytokine production, GF : • ↑ vascular permeability • ↑ procoaggulant activity • ↑ fibroblasts & SM in ECM Complications in blood vessels, kidney, nervous system ….etc Complications are proportional to the degree of hyperglycemia of whatever type 2- The Polyol Pathway Persistent hyperglycemia facilitates entry of glucose & its accumulation into some cells & metabolized into SORBITOL (a polyol) & FRUCTOSE Creation of osmotic gradient Influx of fluid + Toxic lens, retina, peripheral nerves, kidney…etc 3- Activation of Protein Kinase C : • Activation of signal transduction • Leads to production of pro-angiogenic factors (VEGF) Important in retinal neovascularization • Production of pro-fibrogenic factors → ↑ECM & BM thickening COMPLICATIONS Pathology in the Pancreas i -Type I : - Leukocytic infiltration of islets ( T cells) ‘Insulinitis’ with progressive depletion of cells. - Later small indistinct or absent islets. ii - Type II : - Ill defined reduction in islet cell mass - Fibrous tissue accumulation in some islets - Amyloid deposition in islets • Newborn of diabetic mother : islet cell hyperplasia COMPLICATIONS 1- Atherosclerosis : - Cardiovascular - CNS complications - Peripheral circulation 2- Diabetic microangiopathy - Hyaline arteriolosclerosis , exaggerated in hypertension - Diffuse thickening in capillaries of skin, retina peripheral nerves, renal medulla → Leaky vessels→ nephropathy, retinopathy, neuropathy 3- Diabetic nephropathy I - Glomerular lesions- Capillary BM thickening - Nodular glomerulosclerosis 15% -30% ( Kimmelstiel - Wilson lesion) - Diffuse mesangial sclerosis II - Renal vascular lesions - Renal atherosclerosis - Hyaline arteriolosclerosis Kimmelstiel- Wilson lesion Downloaded from: Robbins & Cotran Pathologic Basis of Disease (on 4 December 2005 01:50 PM) © 2005 Elsevier III – Pyelonephritis - Acute & chronic interstitial inflammation - Necrotizing papillitis / papillary necrosis Downloaded from: Robbins & Cotran Pathologic Basis of Disease (on 4 December 2005 01:50 PM) © 2005 Elsevier 4- Ocular complications : I - Retinopathy : - Nonproliferative : hemorrhage, exudate, microaneurysm, edema… - Proliferative : Neovascularization, fibrosis, retinal detachment II - Cataract formation III - Glaucoma 5- Diabetic neuropathy I - Peripheral sensory & autonomic nerve dysfunction ( microangiopathy & demyelination ) II - Neuronal degeneration III - Degenerative spinal cord lesions 6- Recurrent infections : Bacterial & mycotic • Clinical Features in Diabetes : Type 1 : • Age < 20 , but some are latent (LADA) • May present with metabolic acidosis, weight loss, dehydration,& electrolyte imbalance. Polyuria , Polydipsia, Polyphagia ( 3P’s) • Findings : - Hyperglycemia - Glucosuria ± Ketonuria - Electrolyte imbalance Type 2 : • Age > 40yrs., often present incidentally • Patients may have the 3 P’s symptoms of complications • Hyperosmolar nonketotic coma caused by dehydration due to uncompensated hyperglycemic diuresis. • No keto acidosis • Increased susceptibility to infections ISLET CELL TUMORS Islet Cells & Secretions : • • • • β cells insulin α cells glucagon δ cells somatostatin Pancreatic polypeptide ( PP) VIP Islet Cell Tumors of Pancreas : • Include insulinomas, gastrinomas, glucagonomas….etc • Less frequent than pancreatic CA • Maybe functioning or nonfunctioning • Tumors ≤ 2 cm. diameter likely to be benign • Associated clinical syndromes : 1- Hyperinsulinism (Insulinomas) 2- Zollinger - Ellison Syndrome ( Gastrinomas) 3- Multiple endocrine neoplasia (MEN) Insulinoma : • Commonest type • Hypoglycemia ≤ 50 mg./dl. • Attack precipitated by fasting or exercise, relieved by eating or glucose administration • Lab. : serum glucose , serum insulin • Most tumors in pancreas but can be ectopic • Most tumors solitary ( < 2cm.), can be multiple • Majority are benign, 10% can be malignant • Histologically difficult to diagnose malignancy Gastrinomas : • More in middle aged females • Located in pancreas , duodenum or peripancreatic tissue • Single or multiple, or associated with other tumors • > 50% locally invasive or have metastasized at diagnosis • Present with Zollinger- Ellison Syndrome Zollinger - Ellison Syndrome : – Peptic ulcer disease – Ulcer features : Multiple ulcers Unusual locations specially jejunum Intractable – Gastrin hypersecretion – Diarrhea in > 50% & may be the presenting symptom Rare tumors : • α- Cell tumors : Middle aged women Glucagon secretion , mild diabetes, skin rash, anemia • δ- Cell tumors : Somatostatin secretion Diabetes, malabsorption, GB stones… • VIPomas : VIP secretion Watery diarrhea, hypokalemia, achlorhydria ADRENAL GLAND ADRENAL GLAND • Weight of normal gland is 4 gm. Adrenal Cortex - Derived from mesoderm & composed of 1- Zona glomerulosamineralocorticoids (aldosteron) 2- Zona fasciculata glucocorticoids ( cortisol ) 3- Zona reticularis estrogens & androgens • Diseases are those of hyperfunction & hypofunction & tumors Adrenal Medulla – • Derived from neural crest & is part of sympathetic system. • Composed of Chromaffin cells secreting catecholamines • Diseases are mainly tumors Congenital Anomalies • Incidental finding of adrenal tissue in the inguinoscrotal path , mainly in males • Fusion of adrenals • Congenital adrenal hyperplasia • Ectopic tissue in adrenal : liver, thyroid & ovarian tissue ADRENOCORTICAL HYPERFUNCTION : • There are 3 syndromes associated with hyperfunction: 1- Cushing’s Syndrome & Cushing’s Disease 2- Conn’s Syndrome & Hyperaldosteronism 3- Adrenogenital Syndrome CUSHING’Syndrome • Elevation of cortisol level , which occurs in one of four ways A- Endogenous causes : i- ACTH*secreting pituitary microadenoma, few macroadenomas, OR hyperplasia (CUSHING’s DISEASE) ii-Adrenal tumor or hyperplasia iii- Paraneoplastic syndrome B- Exogenous cause : Steroid Therapy Tests used are : Level cortisol in plasma,or excretion of 17hydroxy steroids in urine, diurnal pattern , level of ACTH, & Dexamethasone Suppression test. Morphology of adrenals in Cushing’s Syndrome : • This depends on the cause : 1- Exogenous increase glucocorticoids ACTH Bilateral atrophy of adrenals 2 -Endogenous hypercorticolism: a- Presence of adrenal adenoma or carcinoma, with atrophy of adjacent & contralateral adrenal b- Secondary to ACTH secreting adenoma bilateral diffuse or nodular hyperplasia c- Primary adrenal nodular hyperplasia The pituitary in all forms of Cushing’s syndrome shows Alteration in ACTH producing cells : • Granular basophilic cells show lighter homogenized cytoplasm due to accumulation of intermediate keratin filaments in cytoplasm , called : • Crooke’s Hyaline Change Clinical features of Cushing’s syndrome : Main symptoms include : Central obesity/ moon face Hypertension Hirsutism/ menstrual disturbances Diabetes Osteoporosis Increased risk of infections Pigmentation of skin HYPERALDOSTERONISM : • Excess level of aldosterone cause sodium retension, potassium excretion, resulting in hypertension & hypokalemia. • Type could be primary OR secondary A- Primary : Conn Syndrome • Caused by Adenoma (80%) F:M is 2:1 Single or multiple • Or primary adrenal hyperplasia ( 15% ) , • Carcinoma is rare • Adjacent adrenal cortex is NOT atrophic • There is aldosteron Na retention & K excretion BP , Hypokalemia , RENIN Correctable cause of HYPERTENSION B- Secondary : Due to decreased renal perfusion, activation of the renin - angiotensin system aldosteron • Differentiate from primary by RENIN VIRILIZING Syndromes : Could be caused by - primary gonadal disorders - Adrenocortical Neoplasms - Congenital adrenal hyperplasia • Neoplasms can occur at any age, frequently malignant • Congenital adrenal hyperplasia is caused by an enzyme defect in cortisol synthesis (21 hydroxylase) NO CORTISOLACTH androgenic steroids • Virilization , precocious puberty, ambiguous genitalia • Patients have risk for acute adrenocortical insufficiency Downloaded from: Robbins & Cotran Pathologic Basis of Disease (on 4 December 2005 01:50 PM) © 2005 Elsevier MORPHOLOGY in ALL ADRENAL TUMORS: • Encapsulated , usually yellow • Size variable 1-2 cm. ( 30gms.)Up to large tumors • Most incidental nonfunctioning tumors, may be functioning • Malignant tumors with necrosis, hemorrhage (≥ 300gms) • Usually larger , more aggressive in adults • Both may show same appearance of uniform or slightly pleomorphic cells ,may be eosinophilic or clear • Local invasion ,& the presence of metastases differentiate benign from malignant tumors Cortical Adenoma Downloaded from: Robbins & Cotran Pathologic Basis of Disease (on 4 December 2005 01:50 PM) © 2005 Elsevier Adrenocortical carcinoma ADRENOCORTICAL INSUFFICIENCY : • May be primary adrenal or secondary to destruction of the pituitary as in SHEEHAN’s syndrome….etc • Primary in adrenal may be : A- Acute : 1- Massive adrenal hemorrhage as in anticoaggulant therapy, DIC, sepsis by N.meningitidis,pseudomonas ( Waterhouse- Friderichsen syndrome) 2- Sudden withdrawal of steroid therapy 3- Stress in a pt.with underlying chronic insufficiency Adrenal hemorrhage Downloaded from: Robbins & Cotran Pathologic Basis of Disease (on 4 December 2005 01:50 PM) © 2005 Elsevier Downloaded from: Robbins & Cotran Pathologic Basis of Disease (on 4 December 2005 01:50 PM) © 2005 Elsevier Adrenal insufficiency (continued ) B- Chronic :( Addison’s disease ) Progressive destruction of the adrenal by : 1- Autoimmune Disorder: 75-90 % , may be sporadic or familial, linked to HLA-B8 , DR3, HLA-DQ5 Often multisystem involvement 2- Infections e.g. Tuberculosis , fungii ( AIDS) 3- Metastatic tumors destroying adrenal e.g. lung, breast , …others Morphology & Clinical features in Chronic Adrenal Insufficiency : • Morphology depends on cause : • Autoimmune : Irregular small glands, cortex infiltrated by lymphocytes, medulla normal. • T.B. Caseating Granuloma • Metastatic disease Type of primary tumor • Secondary to pituitary cause : the adrenal is shrunken • Clinical features : Weight loss, hypotension, hypoglycemia, pigmentation…. There is Hyperkalemia & Hyponatremia due to ↓ mineralocorticoids THE ADRENAL MEDULLA : • Composed of CHROMAFFIN CELLS & nerve endings • Secretetes cholamines in response to sympathetic stimulation • Also present in extra-adrenal sites • Pathology includes tumors : A- Pheochromocytoma B- Neuroblastoma PHEOCHROMOCYTOMA : • Secretes catecholamines → VMA • Sometimes described as The 10% Tumor because : * 10% bilateral * 10% extra adrenal ( Paraganglioma) * 10% familial, maybe part of MEN syndrome * 10% Malignant • Usually well circumscribed,small to large in size,maybe pleomorphic. Malignancy confirmed by METASTASES • Clinically sustained or paroxysmal attacks of BP • CORRECTABLE cause of HYPERTENSION Pheochromocytoma Downloaded from: Robbins & Cotran Pathologic Basis of Disease (on 4 December 2005 01:50 PM) © 2005 Elsevier NEUROBLASTOMA : • • • • • • • Commonest extracranial solid tumor of childhood Usually adrenal but maybe extra-adrenal Familial or sporadic Associated with deletion of short arm of chromosome 1 90% associated with catecholamine secretion VMA excreted in 24 hr. urine helpful in diagnosis. Morphologically it is composed of small round blue cells which may differentiate to ganglion cells • Spread to adjacent organs, lymph nodes, renal vein. • Prognosis : STAGE , AGE , N myc amplification MULTIGLANDULAR SYNDROMES POLYGLANDULAR SYNDROME : • Autoimmune disease • Familial or sporadic – Isolated involvement of adrenals – Multiorgan involvement • Type I : autosomal recessive associated with mutation on immune regulator gene on Chr. 21 • Type II : multifactorial, linked to HLA-B8 , HLA-DR3 , HLA-DQ5 – Include Hashimoto’s thyroiditis,adrenalitis, diabetes type I, pernicious anemia MEN SYNDROME : • Inherited syndrome with multiple endocrine tumors & or hyperplasia of component cells • Tumors occur at younger age • Often preceded by asymptomatic OR symptomatic hyperplasia in involved organ • Tumors may be multifocal in the same organ • Often more aggressive than the same tumor without MEN syndrome Types of MEN syndromes : • Type MEN 1 : ( 3 Ps) • Autosomal dominant • Involves suppressor gene on 11q.13 – Parathyroid : multiglandular parathyroid hyperplasia (95%] – Pancreas: aggressive,multifocal functional gastrinomas & insulinomas – Pituitary: Prolactinoma ± GH Type MEN 2 : • Autosomal dominant Proto-oncogen mutation : RET/10q 11 • MEN 2 A : – Medullary carcinoma of thyroid + C cell hyperplasia – Pheochromocytoma (50%) – Parathyroid hyperplasia • MEN 2 B : – As above but no parathyroid hyperplasia – Extra endocrine manifestations : e.g. mucosal neurofibromas