Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Franck–Condon principle wikipedia , lookup
Bose–Einstein condensate wikipedia , lookup
Chemical equilibrium wikipedia , lookup
Temperature wikipedia , lookup
Eigenstate thermalization hypothesis wikipedia , lookup
Chemical thermodynamics wikipedia , lookup
Heat equation wikipedia , lookup
Maximum entropy thermodynamics wikipedia , lookup
Thermodynamics wikipedia , lookup
Transition state theory wikipedia , lookup
Degenerate matter wikipedia , lookup
Heat transfer physics wikipedia , lookup
Work (thermodynamics) wikipedia , lookup
Van der Waals equation wikipedia , lookup
History of thermodynamics wikipedia , lookup
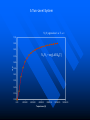
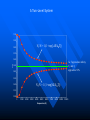
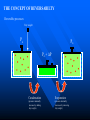
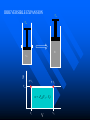
The Distribution of Energy Consider 6 particles – Each particle has three energy levels E3 = 2a a a E E2 = a E1 = 0 Let N = Total number of particles = 6 N1 = Number of particles in state E1 N = N1 + N2 + N3 = 6 N2 = Number of particles in state E2 N2 = Number of particles in state E2 Total energy of the system = 4a N1E1 + N2E2 + N3E3 = 4a The Boltzmann Distribution – Effect of Temperature 1.20 300000K 1.00 Pj = Nj/N 0.80 3000K 0.60 30000K 30K 0.40 300K 0.20 0.00 0 500 1000 1500 2000 2500 Energy 3000 3500 4000 4500 5000 Pj N ,V ,T aj A e E j N ,V / k BT Q N ,V ,T The system partition function is Q N ,V , e E N ,V i qv , N i N! for indistinguishable particles. For the two-level system: N2 e E N1 2 E1 / k B T N1 N 1 1 e E / k BT N2 N 1 1 e E / k BT The Distribution of Molecules on Two Energy Levels Box at rest Box weakly shaken Box vigorously shaken A Two-Level System N2/N1 approaches 1 as T 1.00 0.90 0.80 N2/N1 = exp[-E/kBT] 0.70 N2/N1 0.60 0.50 0.40 0.30 0.20 0.10 0.00 0.00 20000.00 40000.00 60000.00 Tempertaure (K) 80000.00 100000.00 120000.00 A Two-Level System 1.00 0.90 N1/N = 1/(1+exp[-E/kBT]) 0.80 0.70 Ni/N 0.60 As T approaches infinity, N1 and N2 approaches 50% 0.50 0.40 0.30 N2/N = 1/(1+exp[E/kBT]) 0.20 0.10 0.00 0 10000 20000 30000 40000 50000 60000 Temperature (K) 70000 80000 90000 100000 Energy can be evaluated form the partition function: For the system: ln Q ln Q E k BT 2 T N ,V N ,V For the atom/molecule: k BT 2 T N ,V N ,V ln q ln q E N , and for 1 mol: E N A U ln q and so we see that U N A k B T 2 T V Heat capacity and pressure can also be evaluated from the partition function: U Cv and T N ,V ln Q P k BT V N , You should be able to calculate any of these properties for any partition function I give you. Here follows some results for a diatomic molecule. You should understand (and derive completely) how these results were obtained. 3 The translational partition function (3D): 2Mk BT 2 qtrans V ,T V 2 h The rotational partition function (large T): 8 2 Ik BT qrot T h2 The vibrational partition function: e / 2T qvib T 1 e / T vib vib Translational U 3 RT 2 CV 3 R 2 Rotational RT R Vibrational R vib /vib T e 1 2 vib e vib R T 1 e 2 vib /T vib /T 2 Total internal energy is the sum of the individual internal energies and total heat capacity is the sum of the individual heat capacities. Note, we have ignored the electronic contribution. Note q V ,T qtrans qrot qvib qelec and total trans rot vib elec The First Law: Some Terminology System: Well defined part of the universe Surrounding: Universe outside the boundary of the system Heat (q) may flow between system and surroundings Closed system: No exchange of matter with surroundings Isolated System: No exchange of q, w, or matter with surroundings Isothermal process: Temperature of the system stays the same Adiabatic: No heat (q) exchanged between system and surroundings THE CONCEPT OF REVERSABILTY Irreversible processes: Hot Warm Cold Warm Temperature equilibration Mixing of two gases P=0 P=0 Expansion into a vacuum Evaporation into a vacuum THE CONCEPT OF REVERSABILTY Pext = 1 atm Pext = 1 atm REMOVE PINS (1) pins Irreversible Expansion P = 1 atm P = 2 atm Step 2 Step 1 Infinite number of steps (2) Reversible Expansion P = 1 atm P = 2 atm P = 1.999 atm P = 1.998 atm THE CONCEPT OF REVERSABILTY Reversible processes: Tiny weight Po Po Po + P Condensation Evaporation (pressure minimally increases by adding tiny weight) (pressure minimally decreases by removing tiny weight) IRREVERSIBLE EXPANSION Pext Pext V2 V1 P P, V1 P, V2 Pext w = -Pext(V2 – V1) V1 V V2 REVERSIBLE EXPANSION Pext = Pressure of gas. If the gas is ideal, then Pext = nRT/V How does the pressure of an ideal gas vary with volume? This is the reversible path. The pressure at each point along curve is equal to the external pressure. P V V2 V2 V1 V1 w Pext dV PdV V2 1 V2 nRT dV nRT dV nRT ln V 1 V V V1 REVERSIBLE EXPANSION A Pi IRREVERSIBLE EXPANSION The reversible path P A Pi Pext = Pf P B B Pf Pf Vi Vf The shaded area is w nRT ln Vf Vi V Vi The shaded area is w Pf Vi V f Reversible expansion gives the maximum work Vf V REVERSIBLE COMPRESSION IRREVERSIBLE COMPRESSION Pext = Pf B B Pf The reversible path P Pf P A Pi A Pi Vf Vi The shaded area is w nRT ln Vf Vi V Vf Vi The shaded area is w Pf Vi V f Reversible compression gives the minimum work V The Carnot Engine Heat engine Hot reservoir Th Cold reservoir Tc qh qc w q Isothermal expansion P1 V1 T P2 V2 T How can we take it back? w = -nRT ln(V2/V1) The Carnot Engine- what is the efficiency? Isothermal reversible expansion U = 0, w = -nRTh ln(V3/V1) q = +nRTh ln(V3/V1) A Pressure P1 (Th) (Th) B P2 P3 Adiabatic reversible expansion (Tc) dU = CpdT = δw q=0 total work D C (T ) c P4 V1 V2 V3 V4 Adiabatic reversible compression dU = CpdT = δw q=0 Isothermal reversible compression U = 0, w = -nRTc ln(V2/V4) q = +nRTc ln(V2/V4) Statistical Interpretation of Entropy Consider the following expansion: When stopcock is opened: V/2 V/2 At any given moment Probability that a given molecule is in the LHS bulb = 50 % (or ½) Probability that a given molecule is in the RHS bulb = 50 % (or ½) Probability that two molecules stay in LHS bulb = ½ ½ = ¼ (25%) Probability that N molecules stay in LHS bulb = (½)N Microstates: A particular way of arranging molecules among the positions accessible to them while keeping the total energy fixed. Consider four molecules in two compartments: Total number of microstates = 16 1 way 4 ways 6 ways 4 ways 1 way The most probable (the “even split”) If N the “even split” becomes overwhelmingly probable Boltzmann S = kB lnW Consider spin (or dipole restricted to two orientations) 1 particle 2 particles 3 particles 1 mole or , W = 2, and S = kB ln2 , , W = 4, and S = kB ln4 W = 8, and S = kB ln8 W =2NA, and S = kB ln(2NA) = NA kB ln2 = Rln Some entropy problems The spectroscopic entropy of a diatomic molecule is given by the following equation. 2Mk T 3 2 V e 5 / 2 /T S Te B ln ln ln 1 e vib / T vibvib/ T ln g e1 2 R NA rot e 1 h What is meant by residual entropy? (a)Calculate the spectroscopic entropy of CO at 298.15 K and 1 atm. (b)Write down the expression for calculating the (calorimetric) absolute molar entropy from 0 K to 298.15 K. Note that CO undergoes a solid-solid phase transition at 61.6 K. The experimentally determined calorimetric value is ~170 JK -1mol-1.. (c) Calculate the residual entropy of CO. Make the correction for residual to the calorimetric value. Compare the two entropies (spectroscopic and calorimetric). The molar heat capacity of C2H4(g) can be expressed by the following equation over the temperature range 300 K < T < 1000 K. CV T / R 16.4105 6085 .929 K 822826 K 2 T T2 Calculate S if one mole of ethene is heated from 300 K to 600 K at constant volume. There are no phase transitions. Unit 3 – Real & Ideal Gases Non-ideality of gases - Fugacity G P V T G T , P G o T RT ln P P o The thermodynamic function fugacity. Note: f (P,T) P as P 0 1 bar Standard molar Gibbs energy Can generalize for a real gas f P, T f o P P o PV 1 B2 P T P B3 P T P 2 .... RT G T , P G o T RT ln f P, T fo exp B2 P T P B3 P T P 2 ...... Gibbs energy must be taken relative to some standard state Standard state of real gas is taken to be the corresponding ideal gas at 1 bar i.e. must “adjust” the real gas to ideal behavior Real gas (T,P) G2 G1 Ideal gas (T,P) G3 Real gas (T, P 0) = Ideal gas (T, P 0) G1 RT P f ' V dP ' RT ln o RT ln o P f P 0 P P Fugacity coefficient g f Ideal gas, g = 1. Fugacity coefficient measures extent of non-ideality P (Z-1) / P ln g 0 P P Z 1 dP ' ' 0 P P (bar) Real g 1 ideal 0 P (bar) Unit 2 Summary You need to know the Boltzmann equation in terms of probability and fractional occupancy in a two level system. In general, the Boltzmann equation gives the probability, Pj, that a randomly chosen system (from an ensemble of systems in thermal contact) will be in state j with energy Ej(N,V): Pj N ,V ,T aj A e E j N ,V / k BT Q N ,V ,T The system partition function is Q N ,V , e E i N ,V i qv , N N! for indistinguishable particles. For the two-level system: N2 eE N1 2 E1 / k B T , N1 N 1 1 e E / k B T , N2 N 1 1 e E / k BT You should be able to obtain partition functions for simple energy level systems. The Boltzmann equation seeks to find the maximum number of configurations. For a system with large N, there is a configuration with so great a weight that is overwhelms the rest. The system will almost always be found in it, and it will determine the properties of the system. The Boltzmann equation gives the probability of realizing this configuration as a function of energy and temperature. Energy can be evaluated form the partition function: For the system: ln Q ln Q E k BT 2 T N ,V N ,V For the atom/molecule: ln q ln q k BT 2 T N ,V N ,V E N , and for 1 mol: E N A U ln q and so we see that U N A k BT 2 T V Heat capacity and pressure can also be evaluated from the partition function: U and Cv T N ,V ln Q P k BT V N , You should be able to calculate any of these properties for any partition function I give you. Here follows some results for a diatomic molecule. You should understand (and derive completely) how these results were obtained. 3 The translational partition function (3D): 2MkBT 2 qtrans V ,T V h2 The rotational partition function (large T): qrot T 8 2 Ik BT h2 The vibrational partition function: qvib T e / 2T 1 e / T vib vib The vibrational partition function depends on how we define the zero of energy. If we define it as the bottom of the internuclear potential, we obtain the above expression. However if we define zero as the v = 0 state, then we obtain the following expression: e2 e1 = -De v=2 v=1 E = 0 for v = 0 E=0 1/2h qvib T 1 1 e vib / T The expression for the rotational partition function above works for high temperatures. Room temperature measurements have to calculated numerically using the following equation. qrot T 1 3e 2 B / k BT 5e 6 B / k BT From these partition functions the following can be obtained. Translational U 3 RT 2 Rotational RT Vibrational R vib /vib T e 1 2 vib CV 3 R 2 R e R vib T 1 e 2 vib /T vib /T 2 Total internal energy is the sum of the individual internal energies and total heat capacity is the sum of the individual heat capacities. Note, we have ignored the electronic contribution. Note q V ,T qtrans qrot qvib qelec and total trans rot vib elec WORK, ENERGY & HEAT Work is the transfer of energy due to unbalanced forces: w Pext V If Pext is not constant during the process: w Vf Vi For a reversible process (compression or expansion of an ideal gas): V w nRT ln 2 V1 Pext dV The first law of thermodynamics: dU q w Path & state functions: know the U q w difference. Some important processes: ISOLATED SYSTEM: q 0 , w 0 , U 0 ISOTHERMAL (no change in T): U 0 T 0 V wrev q rev nRT ln 2 V1 q 0 ADIABATIC (no change in q): U wrev CV T dT T2 T1 U qV (constant volume) PRESSURE-VOLUME WORK: H U PV constant pressure H U PV q P H The temperature of a gas decreases in a reversible adiabatic expansion: } 3 T2 2 V1 (Know the derivation) V2 T1 ENTHAPLY At constant pressure: H U PV , and q P H To calculate the enthalpy from T = 0K to T = T, must take into account any phase transitions. H T H 0 T fus T CP T dT fus H CP T dT , etc. In other words H CP dT S 0 L T fus Note also that C P H , and C P CV nR T Make sure you know how to calculate enthalpy changes for chemical reactions. You can use Hess’s law and enthalpies of formation to calculate enthalpies of reaction. Know how to calculate enthalpy changes at different temperatures using the following equation: H T2 r H T1 T2 T1 CP products CP react' s ENTROPY Entropy (state function): dS q rev T dS 0 Equilibrium (reversible process) dS 0 Spontaneous process in isolated system Know the Second Law. Isothermal expansion: S R ln V2 , For mixing of two gases: V1 N mix S R yi ln yi (Learn the derivation) j 1 The Third Law: Every substance has finite positive entropy, but at 0 K the entropy may become 0, and does so in the case of a perfectly crystalline substance. Determining absolute entropies from calorimetric data: S T fus C P T dT fus H Tvap C PL T dT vap H T C Pg T dT 0 T fus Tvap T T fus T Tvap T S T These entropies are usually called “calorimetric entropies.” Note we have assumed the Third Law holds true for calorimetric entropies. Statistical Entropy: S k B ln W From partition functions: ln Q qV ,T ln q S k B ln Q k BT Nk B Nk B ln Nk BT T N ,V N T V Note that the above expression relates entropy to the system partition function (Q) and the molecular partition function (q). Make sure you know how to go from the expression for system Q to molecular q, using Stirling’s approximation (lnN! = NlnN – N). The molar entropy of an ideal monoatomic gas: S 3 2mk B T 2 V g e1 5 R R ln h 2 2 NA The molar entropy of a diatomic gas is given by: 3 2Mk BT 2 V e 5 / 2 /T S Te ln ln ln 1 e vib / T vib ln g e1 2 R NA 2 rot e vib / T 1 h You should be able to derive entropy expressions using simple partition functions. You should be able to determine entropies of monoatomic and diatomic molecules from spectroscopic parameters. The entropies are generally called “statistical entropies.” We have assumed the Third Law holds perfectly when calculating calorimetric entropies, i.e. S = 0 at T = 0 K. Spectroscopic entropies are therefore more accurate. Realize that at 0 K some molecules (usually those possessing small dipoles) may get “locked” into two or more possible degenerate states, e.g. CO and linear NNO, W = 2; CH3D, W = 4. You should be able to calculate residual entropies, and add the values to the calorimetric values to get entropies in closer agreement to statistical ones. Helmholtz Energy: A U T S 0 A 0 A 0 A 0 For constant T and V Spontaneous (irreversible) Not spontaneous Equilibrium Gibbs Energy: G H TS G 0 G 0 G 0 For constant T and P Spontaneous (irreversible) Not spontaneous Equilibrium Maxwell Relations Maxwell relations allow us to derive some important thermodynamic equations. You need to be able to derive all of them. Below I have reproduced Table 22.1 from the textbook. Please learn the differential expressions and the derivations for the corresponding Maxwell relations. Thermodynamic Energy Differential expression U dU = TdS – PdV H dH = TdS + VdP A dA = -SdT – PdV G dG = -SdT + VdP Corresponding Maxwell relations T P V S S V T V P S S P S P V T T V S V P T T P Two important ones are the temperature and the pressure dependency of the Gibbs energy. G S This gives: T P proof) G V P T This gives: H G / T 2 Gibbs-Helmholtz equation (learn the T P T G RT ln P2 (ideal gas) P1 P S V , which gives S nR ln 2 (ideal P1 P T T P Other very important one are: gas) A A P and S so that using cross derivatives, we find that V T T V P S V . We end up with S nR ln 2 for an ideal gas. V1 T V V T Beware, I may ask to solve the equations using other equations of state. You need to know how internal energy varies with volume. U P P T (learn the proof) V T T V You need to know how enthalpy varies with pressure. H V V T (learn the proof) P T T P For the above two, you may be asked to solve the partial derivative using an equation of state other than the ideal gas equation (see your homework for examples). The Non-Ideality of Gases At P1 = 1 bar G T , P G o T RT ln P , where G o T = standard molar Gibbs energy. For non-ideal gases G T , P G o T RT ln Using the virial equation for non-ideality: f P ,T fo , where f(P,T) = fugacity of gas. f P ,T P B3 P T P 2 exp B T P ..... 2P o o 2 f P P Z 1 f PV Note, the fugacity coefficient g , and ln g dP , where Z P P RT 0 (Z = compressibility factor) U P 4. The relationship is P T V T T V R P For a van der Waals gas , and so T V V b Integrating U u a U 2 V T V 1 dV which leads to Videal V 2 1 a U ideal U ideal V V V dU a ideal 1 U T ,V a Videal For ethane a = 5.5818 dm6 bar mol-2, and U ideal = 14.55 kJ mol-1 14.53 Molar Energy / kJ/mol Using the van der Waals equation RT a P 2 , we can find P from V b V various molar volumes. We can then determine U as a function of P and plot the two. I have used the molar volume range suggested in problem 22-4. 14.54 14.52 14.51 14.5 14.49 14.48 70 270 470 P /bar R RT P U (a) For an ideal gas and so 0 P V T V V V T a U (b) For a van der Waals gas, see above. 2 V T V RT A (c) For Redlich-Kwong gas, where P 1/ 2 V B T V V B R A P 3/ 2 T V V B 2T V V B 670 R A RT A RT AT U 3/ 2 1/ 2 3/ 2 P V B 2T V V B V B T V V B V B 2T V V B V T A AT A A 1/ 2 3/ 2 1/ 2 1/ 2 T V V B 2T V V B T V V B 2T V V B 2A A 2A A 3A 1/ 2 1/ 2 1/ 2 1/ 2 2T V V B 2T V V B 2T V V B 2T V V B (d) For the virial equation of state Z 1 B2 P T V 1 B3P T V 2 , up to the third virial coefficient. Solving for pressure gives: RT RT RT B2 P T 2 B3 P T 3 V V V R R dB RT R dB RT P B2 P T 2 2 P 2 B3P T 3 3P 3 dT V dT V V V T V V P R dB RT R dB RT R U P T B2 P T 2 2 P 2 B3P T 3 3P 3 dT V dT V V V V T V RT RT RT RT RT dB2 P RT 2 RT dB3P RT 2 B2 P T 2 B3P T 3 B2 P T 2 B3P T 3 V V dT V 2 dT V 3 V V V V dB2 P RT 2 dB3P RT 2 dT V 2 dT V 3 RT 2 dB2 P RT 2 dB3P RT 2 dB2 P 1 dB3P U And so 3 2 dT V dT V V dT V T V 2 dT 5. We can determine how enthalpy varies with P, since H = U + PV. You simply add the corresponding PV values to the U values determined In the last problem. Remember to convert the PV units into joules. Basically 1 L = 1000 cm3 = 10-3 m3, and 1 bar = 105 Pa. Therefore the conversion factor is 102. But we need kJ not J, and so divide further by 1000. The final conversion factor is 0.1. H V We need V T for the remaining parts of the problem. P T T P H (a) For an ideal gas P V RT R T 0 V T P P T T P (b) For the virial equation of state Z 1 B2 P T P B3P T P 2 , up to the third virial coefficient. Solving for volume gives: V RT RT B2 P T RT B3P T RTP B2 P T RT B3 P T RTP P P V T dB R dB B2 P T R 2 P RT B3P T RP 3P RTP dT dT P P H V RT B2 P T RT B3P T RTP V T P P T T P dB dB R T B2 P T R 2 P RT B3P T RP 3P RTP dT dT P dB H dB 2 P RT 2 3P RPT 2 dT dT P T 3.