Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
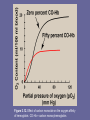
UNIT I: Protein Structure and Function Globular Proteins Overview • This chapter examines relationship b/w structure and function for the clinically important globular hemeproteins, and mainly Hb. II. Globular hemeproteins • Hemeproteins: group of specialized proteins, contain heme as a tightly bound prosthetic group • Role of heme group is dictated by environ. created by 3D structure of protein e.g., – Heme of a cytochrome functions as an electron carrier – Heme of catalase is part of active site of the enzyme catalyzes breakdown of H2O2 – In Hb and myoglobin, the 2 most abundant heme proteins, heme serves to reversibly bind oxygen A. Structure of heme • A complex of protoporphyrin IX and ferrous iron (Fe2+) • Iron is held in center of heme molecule by bonds to 4 nitrogens of porphyrin ring • Heme Fe2+ can form 2 additional bonds, one on each side of the planar porphyrin ring e.g., in myoglobin and Hb, one of these positions is coordinated to side chain of His residue of globin molecule, the other is available to bind oxygen Figure 3.1. A. Hemeprotein (cytochrome c). B. Structure of heme. B. Structure and function of Myoglobin • Myoglobin, a hemeprotein present in heart & skeletal muscle • Functions as a reservoir for oxygen & as oxygen carrier that increases rate of transport of oxygen within muscle cell • Consists of a single polyp structurally similar to individual subunit polyp of Hb molecule, making myoglobin useful model for interpreting some complex properties of Hb 1. • • 2. • • α-helical content: Myoglobin a compact molecule, ~ 80% of its polyp folded into 8 stretches of α-helix These α-helical regions are terminated either by Pro (its 5-membered ring cannot be accommodated in α– helix), or by β-bends and loops stabilized by H-bonds and ionic bonds Location of polar and non-polar aa residues: Interior of myoglobin molecule is composed of almost entirely non-polar aa’s. Packed together forming a structure stabilized by hydrophobic interactions Charged aa’s located almost exclusively on surface, forming H-bonds with each other and with water Figure 3.2. A. Model of myoglobin showing helices A to H. 3. Binding of heme group: – – – – Heme group sits in a crevice lined with non-polar aa’s. Notable exceptions are 2 His residues. One, proximal His, binds directly to iron of heme, 2nd distal His, does not directly interact with heme, but helps stabilize binding of oxygen to ferrous iron The protein, or globin, portion of myoglobin creates a microenviron. for heme that permits reversible binding of one oxygen molecule (oxygenation). Simultaneous loss of electrons by ferrous iron (oxidation) occurs only rarely. B. Schematic diagram of the oxygenbinding site of myoglobin. C. Structure and function of hemoglobin • Hb is found exclusively in RBC’s, its main function is transport of oxygen from lungs to capillaries of tissues. HbA, major Hb in adults, is composed of 4 polyps. 2 αand 2 β-chains –held together by non-covalent interactions. Each subunit has stretches of α-helical structure, and a heme binding pocket. Tetrameric Hb is more complex structurally and functionally than myoglobin e.g., • • • – – – Hb can transport CO2 from tissues to lungs, and carry 4 O2 from lungs to cells of the body, further Oxygen-binding properties of Hb are regulated by interaction with allosteric effectors Figure 3.3 A. Structure of hemoglobin showing the polypeptide backbone. B. Simplified drawing showing the helices. 1. Quaternary structure of Hb • Hb tetramer can be envisioned as composed of 2 identical dimers (αβ)1 and (αβ)2. • The 2 polyp chains in each dimer held tightly together primarily by hydrophobic interactions (in this case hydrophobic aa residues are localized not only in interior of molecule but also in a region on surface of each subunit. Interchain hydrophobic interactions form strong associations b/w α- and β-subunits in dimers) • Ionic and H-bonds also occur b/w members of the dimer • The two dimers, in contrast, are able to move wrt each other, being held primarily by polar bonds. The weaker interactions b/w these mobile dimers result in the 2 dimers occupying different relative positions in deoxy-Hb as compared with oxy-Hb Figure 3.4. Schematic diagram showing structural changes resulting from oxygenation and deoxygenation of hemoglobin. a) T-form: • • • deoxy form of Hb “T”, or taut (tense) form. The 2 αβ dimers interact through a network of ionic and H-bonds constrain movement of polyp chains Low oxygen-affinity form of Hb b) R-form: – – – Binding of oxygen to Hb causes rupture of some of the ionic and H-bonds b/w αβ dimers This leads to a structure called “R” or relaxed form, here polyp chains have more freedom of movement R-form is high oxygen-affinity form of Hb D. Binding of oxygen to myoglobin and Hb • • • Myoglobin can bind 1 O2 molecule, it contains only 1 heme group Hb can bind 4 O2 molecules, one at each of its 4 heme groups Degree of saturation (Y) of these oxygenbinding sites on all myoglobin or Hb molecules can vary b/w zero (all sites are empty) and 100% (all sites are full) 1. Oxygen dissociation curve • A plot of Y measured at different pO2 • Curves for myoglobin & Hb show important differences. Myoglobin has a higher oxygen affinity than Hb. Partial pressure of oxygen needed to achieve half-saturation of binding sites (P50) is ~ 1mm Hg for myoglobin & 26 mm Hg for Hb • Note: the higher the oxygen affinity (i.e., the more tightly oxygen binds), the lower P50 Figure 3.5 Oxygen dissociation curves for myoglobin and hemoglobin. a. Myoglobin • • • The oxygen dissociation curve for myoglobin has a hyperbolic shape. This reflects myoglobin reversibly binds a single molecule of oxygen Thus, oxygenated (MbO2) and deoxygenated (Mb) exist in a simple equilibrium: Mb + O2 ↔ MbO2 Mb is designed to bind oxygen released by Hb at the low pO2 found in muscles. Mb releases oxygen within muscle cell in response to oxygen demand b. Hb • • • The oxygen dissociation curve for Hb is sigmoidal in shape. This reflects that subunits cooperate in binding oxygen. Cooperative binding of O2 by the 4 subunits of Hb means binding of O2 to one heme group increases the oxygen affinity of remaining heme groups in the same Hb molecule, this effect is heme-heme interaction Although binding of 1st O2 is difficult, subsequent binding of O2 occurs with high affinity, shown by steep upward curve in the region 20-30 mm Hg Figure 3.6 Hemoglobin binds oxygen with increasing affinity. E. Allosteric effects • Ability of Hb to reversibly bind oxygen is affected by pO2 (through heme-heme interaction), pH of environ. pCO2 and availability of 2,3-bisphosphoglycerate. • Collectively called allosteric (“other site”) effectors, as their interaction at one site on Hb molecule affects binding of oxygen to heme groups at other locations on the molecule • Binding of oxygen to myoglobin is not influenced by allosteric effectors of Hb 1. Heme-heme interactions: sigmoidal oxygen-binding curve reflects specific structural changes that are initiated at one heme and transmitted to other heme groups in Hb tetramer. Net effect affinity of Hb for last oxygen ~ 300x greater than affinity for 1st oxygen a. Loading and unloading oxygen: cooperative binding of oxygen allows Hb deliver more oxygen to tissues in response to relatively small changes in pO2. Figure 3.5 indicates pO2 in alveoli of lung and capillaries of tissues - e.g., in lung conc. oxygen is high and Hb becomes saturated (loaded) with oxygen. - in peripheral tissues, oxy-Hb releases (unloads) much of its oxygen for use in oxidative metabolism b. Significance of sigmoidal O2-dissociation curve: Steep slope of O2dissociation curve over the range of oxygen conc. b/w lungs and tissues permits Hb to carry and deliver oxygen efficiently from sites of high to sites of low pO2 A molecule with hyperbolic O2-dissociation curve, e.g. myoglobin could not achieve the same thing. Instead, it would have max affinity for oxygen throughout this oxygen pressure would deliver no oxygen to tissues Figure 3.7. Transport of oxygen and CO2 by hemoglobin. 2. Bohr effect: – – – Release of oxygen from Hb is enhanced when pH is lowered or when Hb is in pressure of an increased pCO2. Both result in decreased oxygen affinity shift to the right in O2-dissociation curve. This change in oxygen binding is called Bohr effect. Conversely, raising pH or lowering conc. of CO2 greater affinity for oxygen, and a shift to the left in O2-dissociation curve. Figure 3.8. Effect of pH on the oxygen affinity of hemoglobin. a. Source of the protons that lower the pH: • Conc. of both CO2 and H+ in capillaries of metabolically active tissues is higher than that observed in capillaries of lung, where CO2 is released into expired air • Note: organic acids e.g., lactic acid, are produced during anaerobic metabolism in rapidly contracting muscle • In tissues, CO2 is converted by carbonic anhydrase to carbonic acid, CO2 + H2O ↔ H2CO3 which spontaneously loses a proton becoming bicarbonate, the major blood buffer H2CO3 ↔ H+ + HCO3• The proton produced contributes to lowering pH. This differential pH gradient (lungs having higher, tissues lower pH) favors unloading oxygen in peripheral tissues, and loading of oxygen in lung. • Thus, oxygen affinity of Hb responds to small shifts in pH b/w lungs and oxygen-consuming tissues, making Hb a more efficient transporter of oxygen. b. Mechanism of the Bohr effect: Deoxy form of Hb has a greater affinity for protons than does oxyHb. This fact is caused by ionizable groups, e.g., N-terminal αamino groups, & specific His side chains that have higher pKa’s in deoxy-Hb than in oxy-Hb. An increase in conc. of protons causes these groups to become protonated (charged) and able to form ionic bonds (a.k.a salt bridges), which stabilize deoxy form of Hb, producing a decrease in oxygen affinity Bohr effect schematically: HbO2 (oxy-Hb) + H+ ↔ HbH (deoxy-Hb) + O2 where an increase in protons (or a lower pO2) shifts equilibrium to right, whereas an increase in pO2 (or decrease in protons) shifts equilibrium to left 3. Effect of 2,3 bisphosphoglycerate on oxygen affinity • 2,3 BPG an important regulator of binding of oxygen to Hb • It is the most abundant organic phosphate in RBC, where its conc. ~ that of Hb. • 2,3 BPG is synthesized from an intermediate of glycolytic pathway Figure 3.9. Synthesis of 2,3-BPG. [Note: is a phosphoryl group.] a. Binding of 2,3 BPG to deoxy-Hb 2,3 BPG decreases oxygen affinity of Hb by binding to deoxy-Hb but not to oxy-Hb. This preferential binding stabilizes the “taut” conformation of deoxy-Hb. HbO2 + 2,3-BPG ↔ Hb-2,3-BPG + O2 b. Binding site of 2,3 BPG One molecule of 2,3 BPG binds to a pocket, formed by the two β-globin chains, in center of deoxy-Hb tetramer. This pocket contains several positively charged aa’s that form ionic bonds with the negatively charged phosphate groups of 2,3 BPG A mutation of one of these residues can result in Hb variants with abnormally high oxygen affinity 2,3 BPG is expelled on oxygenation of Hb. Figure 3.10. Binding of 2,3-BPG by deoxyhemoglobin. c. Shift of oxygen dissociation curve • Hb from which 2,3 BPG removed, has a high affinity for oxygen. In RBC, presence of 2,3 BPG significantly reduces affinity of Hb for oxygen, shifting oxygendissociation curve to the right. This reduced affinity enables Hb to release oxygen efficiently at partial pressures found in tissues Figure 3.11. Effect of 2,3-BPG on the oxygen affinity of hemoglobin. d. Response of 2,3-BPG levels to chronic hypoxia or anemia • Conc. of 2,3 BPG in RBC increases in response to chronic hypoxia, e.g., that observed in obstructive pulmonary emphysema, or at high altitudes, where Hb may have difficulty receiving sufficient oxygen. Intracellular 2,3 BPG also elevated in chronic anemia in which fewer than normal RBCs are available to supply body’s oxygen needs. Elevated 2,3 BPG levels lower oxygen affinity of HB, permitting greater unloading of oxygen in capillaries of tissues e. Role of 2,3 BPG in transfused blood: • 2,3 BPG is essential for normal oxygen transport function of Hb. • e.g., storing blood in acid-citrate-dextrose decrease of 2,3 BPG in RBCs. Such blood displays an abnormally high oxygen affinity, and fails to unload its bound oxygen properly in the tissues. • Hb deficient in 2,3 BPG thus acts as an oxygen “trap” rather than as an oxygen transport system. • Transfused RBCs are able to restore depleted supplies of 2,3 BPG in 24-48 h. However, severely ill patients may be seriously compromised if transfused with large quantities of such 2,3 BPG –”stripped” blood. • Decrease in 2,3 BPG can be prevented by adding substrates e.g., inosine (hypoxanthine-ribose) to storage medium. • Inosine, uncharged molecule can enter RBC, its ribose moiety is released, phosphorylated, and enters hexose mosophosphate pathway, eventually converted to 2,3 BPG 4. Binding of CO2: • Most CO2 produced in metabolism is hydrated and transported as bicarbonate ion. • However, some CO2 is carried as carbamate bound to uncharged α-amino groups of Hb (carbamino-Hb): Hb-NH2 + CO2 ↔ Hb-NH-COO- + H+ • Binding of CO2 stabilizes T (taut) or deoxy form of Hb, resulting in decrease in its affinity for oxygen. In lungs, CO2 dissociates from Hb, released in breath. 5. Binding of CO: • CO binds tightly (but reversibly) to Hb iron carbon monoxyhemoglobin (HbCO). • When CO binds to one or more of the 4 heme sites, Hb shifts to relaxed conformation, causing remaining sites bind oxygen with high affinity. • This shifts oxygen saturation curve to the left, and changes normal sigmoidal shape toward a hyperbola. • As a result affected Hb is unable to release oxygen to tissues • Affinity of Hb to CO is 220x greater than for oxygen. Figure 3.12. Effect of carbon monoxide on the oxygen affinity of hemoglobin. CO-Hb = carbon monoxyhemoglobin. • Even minute quantities of CO in environ. can produce toxic conc’s of HbCO in blood. • CO toxicity appears to result from a combination of tissue hypoxia and direct COmediated damage at cellular level. • CO poisoning is treated with 100% oxygen therapy, which facilitates dissociation of CO from Hb. F. Minor hemoglobins • HbA is one member of a functionally & structurally related family of proteins, the hemoglobins • Each of these oxygen-carrying proteins is a tetramer, composed of 2 α-like and 2 ß-like polyps • Certain Hbs e.g., HbF, are normally synthesized only during fetal development, others e.g., HbA2, are synthesized in the adult although at low levels compared to HbA • HbA can also become modified by covalent addition of a hexose e.g., addition of glucose glucosylated Hb derivative HbA1c. Figure 3.13 Normal adult human hemoglobins. [Note: The a-chains in these hemoglobins are identical.] 1. Fetal hemoglobin (HbF): • HbF is a tetramer of two α, plus 2 γ chains (α2γ2). γ chains are members of ß-globin gene family. a. HbF synthesis during development: – – – – In 1st few wks after conception, embryonic Hb (Hb Gower 1), composed of 2 zeta and 2 epsilon chains (ζ2ε2), synthesized by embryonic yolk sac Within few wks, fetal liver begins to synthesize HbF in developing bone marrow HbF is major Hb found in fetus and newborn, accounting for ~60% of total Hb in RBCs during last months of fetal life HbA synthesis starts in BM at about 8th month of pregnancy and gradually replaces HbF Figure 3.14 Developmental changes in hemoglobin. b. Binding of 2,3 BPG to HbF: - Under physiologic conditions, HbF has a higher affinity for oxygen than does HbA, as a result HbF’s binding only weakly to 2,3 BPG - The γ-globin chains lack some of the positively charged aa’s responsible for binding 2,3 BPG in ß-globin chains - Because 2,3 BPG serves to reduce affinity of HbA for oxygen, weaker interaction b/w 2,3 BPG and HbF higher oxygen affinity for HbF relative to HbA. - If both HbF and HbA stripped of their 2,3 BPG they then have similar affinity for oxygen - The higher oxygen affinity of HbF facilitates transfer of oxygen from maternal circulation across placenta to RBCs of fetus. HbA2: • Is a minor component of normal adult Hb, appearing ~ 12 wks after birth ~ 2% of total Hb. composed of α2δ2. HbA1c: • Under physiologic conditions, HbA slowly & non-enzymatically glycosylated • Extent of glycosylation dependent on plasma conc. of a particular hexose • Most abundant form of glycosylated Hb is HbA1c: has glucose residues attached predominantly to NH2 groups of the N-terminal Val of ß-globin chains • Increased amounts found in RBCs of diabetes mellitus, as their HbA has contact with higher glucose concs during the 120 d lifetime of these cells Figure 3.15 Nonenzymic addition of glucose to hemoglobin. III. Organization of the globin genes To understand diseases resulting from genetic alterations in structure or synthesis of Hbs. A. α-Gene family Genes for α- and β-globin-like subunits of Hb chains occur in 2 separate gene clusters (families) located on two different chr’s α-gene cluster on chr 16 contains 2 genes for αglobin chains. It also contains zeta (ζ) gene, expressed early in development as a component of embryonic Hb, & a number of globin-like genes that are not expressed (pseudogenes) B. β-Gene family - A single β-globin gene located on chr 11, plus additional 4 β-globin-like genes: epsilon (ε) gene, two gamma (γ) genes (Gγ & Aγ that are expressed in fetal, HbF), and the δ gene that codes for globin chain found in minor adult HbA2. Figure 3.16 Organization of the globin gene families. C. Steps in globin chain synthesis • Begins in nucleus of RBC precursors, where DNA sequences encoding gene is transcribed. • RNA produced by transcription is a precursor of mRNA • Before translation, two non-coding stretches of RNA (introns) must be removed, and remaining 3 fragments (exons) reattached in a linear manner. • Resulting mature mRNA enters cytosol, its genetic info. translated globin chain Figure 3.17 Synthesis of globin chains. IV. Hemoglobinopathies • Family of disorders caused by production of structurally abnormal Hb, synthesis of insufficient quantities of normal Hb, or rarely both. • Sickle-cell anemia (HbS), Hb C disease (HbC), and thalassemia syndromes are examples that can have severe clinical consequences. • HbS & HbC result from altered aa sequences, thalassemia caused by decreased production A. Sickle cell disease (HbS disease) • a.k.a sickle cell anemia, a genetic disorder of blood, caused by a single nt alteration (point mutation) in β-globin gene. • Most common inherited blood disorder in US, affecting ~80,000 Americans (primarily, AfricanAmerican population, affecting 1/500 newborn infants) • Homozygous recessive disorder. Occurs in individuals inherited 2 mutant genes that code for β-chains • Mutant β-globin chain = βS, resulting Hb = α2βS2 HbS • Sickle cell disease is characterized by lifelong episodes of pain (“crises”), chronic hemolytic anemia, and increased susceptibility to infections, usually beginning in early childhood. • Other symptoms include: acute chest syndrome, stroke, splenic & renal dysfunction • Lifetime of an erythrocyte homozygous for HbS is ~20 days, compared with 120 d for normal RBCs • Heterozygotes (1 of 10 African-Americans, have one normal and one sickle cell gene). Their RBCs contain both HbS and HbA. These individuals have sickle cell trait usually do not show clinical symptoms & can have normal life span Figure 3.18. Sickle cell disease in the United States. Figure 3.19 Amino acid substitutions in HbS and HbC. Figure 3.20. A. Photograph of a gel prior to electrophoresis. B. Diagram of hemoglobins A, S, and C after electrophoresis. 2. Sickling causes tissue anoxia: - Substitution of non-polar Val for a charged Glu forms a protrusion on the β-globin that fits into a complementary site on the α-chain of another Hb molecule in the cell. - At low oxygen tension, HbS polymerizes inside RBCs 1st forming a gel, then assembling into a network of fibrous polymers that stiffen and distort cell, producing rigid, misshapen RBCs. - Such sickled cells frequently block flow of blood in narrow capillaries. This interruption in supply of oxygen localized anoxia (oxygen deprivation) in the tissue pain and eventually death (infarction) of cells in vicinity of blockage. Figure 3.21 Molecular and cellular events leading to sickle cell crisis. 3. Variables that increase sickling: - Extent of sickling & severity of disease is enhanced by any variable that increases proportion of HbS in deoxy state (i.e., reduces affinity of HbS for oxygen) - These variables include decreased oxygen tension as a result of high altitude or flying in non-pressurized plane, increased pCO2, decreased pH, and an increased conc. of 2,3 BPG in RBCs 4. Treatment: Involves adequate hydration, analgesics, aggressive antibiotic therapy if infection present, & transfusions in patients at high risk for fatal vasocclusions. Intermittent transfusions with packed RBCs reduce risk of stroke, but benefits must be weighed against complications of transfusion, which include iron overload (hemosiderosis), blood-borne infections, and immunologic complications. Hydroxyurea (an anti-tumor drug) decreases frequency of painful crises & reduces mortality. Mechanism of action not fully understood, but may include a modest increase in fetal Hb, which decreases sickling 5. Possible selective advantage of heterozygous state: High freq of HbS gene among black Africans, despite damaging effect in homozygous state, suggests that a selective advantage for heterozygous individuals E.g., heterozygotes for sickle cell gene are less susceptible to malaria, caused by Plasmodium falciparum, which spends part of its life cycle in RBC. As RBCs in heterozygotes, like those in homozygotes, have shorter lifespan than normal, parasite cannot complete the intracellular stage of its development. This may provide selective advantage to heterozygotes living in regions where malaria is a major cause of death. Figure 3.22 A. Distribution of sickle cell in Africa expressed as percentage of the population with disease. B. Distribution of malaria in Africa. B. Hemoglobin C disease • Also has a single aa substitution in 6th position of β-globin chain; a Lys is substituted for Glu • This substitution HbC to move more slowly toward anode than does HbA or HbS • Patients homozygous for HbC generally have mild, chronic hemolytc anemia. Do not suffer from infarctive crises, and no specific therapy is required C. Hemoglobin SC disease • Some β-globin chains have sickle cell mutation, other βglobin carry mutation found in HbC disease (compound heterozygotes) • Hb levels tend to be higher in HbSC disease than in sickle cell disease, and may even be at low end of normal range • Clinical course of adults with HbSC disease differs from that of sickle cell disease, in which patients generally have painful crises beginning in childhood • It is common for patients with HbSC to remain well (and undiagnosed) until they suffer infarctive crisis. This crisis often follows childbirth or surgery and may be fatal. D. Methemoglobinemias • Oxidation of heme component of Hb to ferric (Fe3+) state forms methemoglobin, which cannot bind oxygen • This oxidation may be caused by action of certain drugs, e.g., nitrates, or endogenous products, e.g., reactive oxygen intermediates • The oxidation may also result from inherited defects, e.g., certain mutations in α- or β-globin chain promote formation of methemoglobin (HbM) • Further, deficiency of NADH-cytochrome b5 reductase (a.k.a NADH methemoglobin reductase), enzyme responsible for conversion of methemoglobin (Fe3+) to Hb (Fe2+) accumulation of HbM • RBCs of newborns have ~one-half capacity of adults to reduce HbM. They are particularly susceptible to effects of HbM-producing cpds • Methemoglobinemias are characterized by “chocolate cyanosis” (a brownish blue coloration of skin and membranes) & chocolate coloredblood, as a result of dark-colored HbM. • Symptoms are related to tissue hypoxia, and include anxiety, headache, and dyspnea. In rare cases, coma & death can occur. E. Thalassemias • Hereditary hemolytic diseases, imbalance in synthesis of globin chains • As a group, most common single-gene disorders in humans • Normally, synthesis of β- and α-globin chains are coordinated, so each has a partner formation of α2β2 (HbA). In thalassemia synthesis of either chain is defective • Can be caused by a variety of mutations, including entire gene deletions, substitution or deletion of one to many nt’s in DNA • Each thalassemia can be classified as a disorder with no globin chains produced (βº- or αº-thalassemia) or one in which some synthesized, but at reduced rate (β+- or α+thalassemia) β-Thalassemia: • Synthesis of β-globin chains decreased or absent, whereas α-globin chains synthesis is normal. • α-globin chains cannot form stable tetramers and precipitate premature death of cells initially destined to become mature RBCs • Accumulation of α2γ2 (HbF) and γ4 (Hb Bart’s) also occurs • There are only 2 copies of β-globin genes individuals with β-globin gene defects have either β-thalassemia trait (β-thalassemia minor) if only one copy defective, or β-thalassemia major if both genes are defective • As β-globin gene is not expressed until late in fetal gestation, physical manifestations of β-thalassemia appear only after birth. • Individuals with β-thalassemia minor make some βglobin chains and usually do not require specific treatment. • However, infants born with β-thalassemia major seem healthy at birth, but become severely anemic, usually during 1st or 2nd yr of life. These patients require regular transfusions of blood • Although this treatment is lifesaving, cumulative effect of transfusions is iron overload (a syndrome known hemosiderosis) which typically causes death b/w ages 15-25 yrs • Increasing use of BM replacement therapy has been a boon to these patients Figure 3.23 A. β-Globin gene deletions in the βthalassemias. B. Hemoglobin tetramers formed in βthalassemias. α-Thalassemia: • Synthesis of α-globin chains decreased or absent. • As each individual’s genome contains 4 copies of α-globin gene, there are several levels of α-globin chain deficiencies • If 1 of the 4 genes is defective, individual is silent carrier of αthalassemia no physical manifestations of disease occur • If 2 α-globin genes are defective α-thalassemia trait • If 3 α-globin genes are defective individual has hemoglobin H (HbH) disease –a mildly to moderately severe hemolytic anemia • Synthesis of unaffected γ- and then β-globin chains continues accumulation of γ tetramers in newborn (γ4, Hb Bart’s) or β tetramers (β4, HbH) • Although these tetramers are fairly soluble, the subunits show no heme-heme interaction. Their oxygen dissociation curves are almost hyperbolic they have very high oxygen affinities essentially useless as oxygen deliverers to tissues • If all 4 α-globin genes are defective, hydrops fetalis and fetal death result, because α-globin chains are required for synthesis of HbF Figure 3.24. A. α-Globin gene deletions in the α-thalassemias. B. Hemoglobin tetramers formed in αthalassemias. Summary • HbA major adult Hb : α2ß2 held together by noncovalent interactions. Subunits occupy different relative positions in deoxy- & oxyhemoglobin • Deoxy- form = “T” or taut (tense) form. It has a constrained structure that limits movement of polyp chains • T form is low oxygen affinity form. • Binding of oxygen rupture of some ionic & H-bonds R (relaxed form), subunits have more freedom of movement • R form is high oxygen affinity form • Oxygen dissociation curve for Hb is sigmoidal (that of myoglobin is hyperbolic) indicating subunits cooperate in binding oxygen i.e., binding to one heme increases affinity of remaining heme groups in same Hb molecule • Hb’s ability to bind oxygen reversibly is affected by pO2, and availability of 2,3 BPG e.g., release of O2 from Hb is enhance when pH lowered or oCO2 increased (Bohr effect), e.g., in exercising muscle, & oxygen dissociation curve of Hb is shifted to right. • To cope with long-term effects of chronic hypoxia or anemia, conc of 2,3 BPG in RBCs increases. 2,3 BPG binds Hb and decreases affinity to oxygen shifts curve to right • CO binds tightly (but reversibly) to Hb iron, carbon monoxyHb (HbCO) • Hemoglobinopathies are disorders of Hb caused by production of structurally abnormal or synthesis of insufficient quantities of normal Hb subunits, or rarely, both. • HbS disease, HbC disease, & thalassemia syndromes are examples and can have severe clinical consequences Figure 3.25. Key concept map for hemoglobin structure and function. Figure 3.26. Key concept map for hemoglobinopathies.