Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
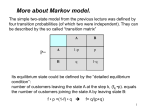
Biological Sequences and Hidden Markov Models CBPS7711 Sept 9, 2010 Sonia Leach, PhD Assistant Professor Center for Genes, Environment, and Health National Jewish Health [email protected] Slides created from David Pollock’s slides from last year 7711 and current reading list from CBPS711 website Center for Genes, Environment, and Health Introduction • Despite complex 3-D structure, biological molecules have primary linear sequence (DNA, RNA, protein) or have linear sequence of features (CpG islands, models of exons, introns, regulatory regions, genes) • Hidden Markov Models (HMMs) are probabilistic models for processes which transition through a discrete set of states, each emitting a symbol (probabilistic finite state machine) • HMMs exhibit the ‘Markov property:’ the conditional probability distribution of future states of the process depends only upon the present state (memory-less) • Linear sequence of molecules/features is Andrey Markov modelled as a path through states of the HMM 1856-1922 which emit the sequence of molecules/features • Actual state is hidden and observed only through output symbols Center for Genes, Environment, and Health 2 Hidden Markov Model • Finite set of N states X • Finite set of M observations O • Parameter set λ = (A, B, π) – Initial state distribution πi = Pr(X1 = i) – Transition probability aij = Pr(Xt=j | Xt-1 = i) – Emission probability bik = Pr(Ot=k | Xt = i) Example: 1 2 3 N=3, M=2 π=(0.25, 0.55, 0.2) A = 0 0.2 0.8 B = 0.1 0 0.9 0.1 1.0 0 0 Center for Genes, Environment, and Health 0.9 0.75 0.25 0.5 0.5 3 Hidden Markov Model • Finite set of N states X • Finite set of M observations O • Parameter set λ = (A, B, π) Hidden Markov Model (HMM) X t-1 Xt Ot-1 Ot – Initial state distribution πi = Pr(X1 = i) – Transition probability aij = Pr(Xt=j | Xt-1 = i) – Emission probability bik = Pr(Ot=k | Xt = i) Example: 1 2 3 N=3, M=2 π=(0.25, 0.55, 0.2) A = 0 0.2 0.8 B = 0.1 0 0.9 0.1 1.0 0 0 Center for Genes, Environment, and Health 0.9 0.75 0.25 0.5 0.5 4 Probabilistic Graphical Models Y X Time Markov Process (MP) Xt− 1 Xt Observability Hidden Markov Model (HMM) X t-1 Xt Ot-1 Ot A t− 1 Markov Decision Process X t − 1 (MDP) U t− 1 Center for Genes, Environment, and Health At Utility Partially Observable X t Markov Decision Process U (POMDP) t Observability and Utility A t− 1 Ot− 1 At Xt− 1 Xt U t− 1 Ut Ot 5 Three basic problems of HMMs 1. Given the observation sequence O=O1,O2,…,On, how do we compute Pr(O| λ)? 2. Given the observation sequence, how do we choose the corresponding state sequence X=X1,X2,…,Xn which is optimal? 3. How do we adjust the model parameters to maximize Pr(O| λ)? Center for Genes, Environment, and Health 6 Example: 1 πi = Pr(X1 = i) aij = Pr(Xt=j | Xt-1 = i) bik = Pr(Ot=k | Xt = i) 2 3 N=3, M=2 π=(0.25, 0.55, 0.2) A = 0 0.2 0.8 B = 0.1 0 0.9 0.1 1.0 0 0 0.9 0.75 0.25 0.5 0.5 • Probability of O is sum over all state sequences Pr(O|λ) = ∑all X Pr(O|X, λ) Pr(X|λ) = ∑all X πx1 bx1o1 ax1x2 bx2o2 . . . axn-1xn bxnon • Efficient dynamic programming algorithm to do this: Forward algorithm (Baum and Welch) Center for Genes, Environment, and Health 7 A Simple HMM CpG Islands where in one state, much higher probability to be C or G 0.8 G C A T .3 .3 .2 .2 CpG 0.9 0.2 0.1 G C A T .1 .1 .4 .4 Non-CpG From David Pollock CpG G C A T The Forward Algorithm 0.8 .3 .3 .2 .2 0.2 G C A T Probability of a Sequence is the Sum of All Paths that Can Produce It G .3 0.1 .1 .1 .4 .4 0.9 G .1 Pr(G|λ) = πC bCG + πN bNG = .5*.3 + .5*.1 For convenience, let’s drop the 0.5 for now and add it in later G Adapted from David Pollock’s Non-CpG CpG G C A T The Forward Algorithm 0.8 .3 .3 .2 .2 0.2 G C A T Probability of a Sequence is the Sum of All Paths that Can Produce It G .3 0.1 .1 .1 .4 .4 0.9 (.3*.8+ .1*.1)* .3 =.075 G .1 (.3*.2+ .1*.9)* .1 =.015 G C For O=GC have 4 possible state sequences CC,NC, CN,NN Adapted from David Pollock’s Non-CpG CpG G C A T The Forward Algorithm 0.8 .3 .3 .2 .2 0.2 G C A T Probability of a Sequence is the Sum of All Paths that Can Produce It (.3*.8+ .1*.1) *.3 =.075 (.075*.8+ G .1 (.3*.2+ .1*.9) *.1 =.015 (.075*.2+ .015*.9) *.1 =.0029 G C G G .3 0.1 .1 .1 .4 .4 0.9 .015*.1) *.3 =.0185 For O=GCG have possible state sequences CCC, CCN NCC, NCN NNC, NNN CNC, CNN Adapted from David Pollock’s Non-CpG CpG G C A T The Forward Algorithm 0.8 .3 .3 .2 .2 0.2 G C A T Probability of a Sequence is the Sum of All Paths that Can Produce It (.3*.8+ .1*.1) *.3 =.075 (.075*.8+ G .1 (.3*.2+ .1*.9) *.1 =.015 (.075*.2+ .015*.9) *.1 =.0029 G C G G .3 0.1 .1 .1 .4 .4 0.9 .015*.1) *.3 =.0185 For O=GCG have possible state sequences CCC, CCN NCC, NCN NNC, NNN CNC, CNN Adapted from David Pollock’s Non-CpG CpG G C A T The Forward Algorithm 0.8 .3 .3 .2 .2 0.2 G C A T Probability of a Sequence is the Sum of All Paths that Can Produce It G .3 0.1 .1 .1 .4 .4 0.9 G .1 G (.3*.8+ .1*.1) *.3 =.075 (.075*.8+ (.0185*.8 (.003*.8+ +.0029*.1 .0025*.1) *.3 =.0185 )*.2 =.003 *.2 =.0005 (.3*.2+ .1*.9) *.1 =.015 (.075*.2+ .015*.9) *.1 =.0029 (.0185*.2 *.4 =.0025 (.003*.2+| .0025*.9) *.4 =.0011 C G A A .015*.1) +.0029*.9) Adapted from David Pollock’s Non-CpG CpG G C A T The Forward Algorithm 0.8 .3 .3 .2 .2 0.2 G C A T Probability of a Sequence is the Sum of All Paths that Can Produce It G .3 0.1 .1 .1 .4 .4 0.9 Non-CpG G .1 (.3*.8+ .1*.1) *.3 =.075 (.075*.8+ (.3*.2+ .1*.9) *.1 =.015 (.0185*.8 (.003*.8+ +.0029*.1 .0025*.1) *.3 =.0185 )*.2 =.003 *.2 =.0005 (.075*.2+ .015*.9) *.1 =.0029 (.0185*.2 (.003*.2+| .0025*.9) *.4 =.0011 .015*.1) +.0029*.9) *.4 =.0025 G C G A A Problem 1: Pr(O| λ)=0.5*.0005 + 0.5*.0011= 8e-4 CpG G C A T The Forward Algorithm 0.8 .3 .3 .2 .2 0.2 G C A T Probability of a Sequence is the Sum of All Paths that Can Produce It G .3 0.1 .1 .1 .4 .4 0.9 Non-CpG G .1 (.3*.8+ .1*.1) *.3 =.075 (.075*.8+ (.3*.2+ .1*.9) *.1 =.015 (.0185*.8 (.003*.8+ +.0029*.1 .0025*.1) *.3 =.0185 )*.2 =.003 *.2 =.0005 (.075*.2+ .015*.9) *.1 =.0029 (.0185*.2 (.003*.2+| .0025*.9) *.4 =.0011 .015*.1) +.0029*.9) *.4 =.0025 G C G A A Problem 2: What is optimal state sequence? CpG G C A T The Forward Algorithm 0.8 .3 .3 .2 .2 0.2 G C A T Probability of a Sequence is the Sum of All Paths that Can Produce It G .3 0.1 .1 .1 .4 .4 0.9 G .1 G (.3*.8+ .1*.1) *.3 =.075 (.075*.8+ (.0185*.8 (.003*.8+ +.0029*.1 .0025*.1) *.3 =.0185 )*.2 =.003 *.2 =.0005 (.3*.2+ .1*.9) *.1 =.015 (.075*.2+ .015*.9) *.1 =.0029 (.0185*.2 *.4 =.0025 (.003*.2+| .0025*.9) *.4 =.0011 C G A A .015*.1) +.0029*.9) Adapted from David Pollock’s Non-CpG CpG G C A T 0.8 .3 .3 .2 .2 0.2 G C A T The Viterbi Algorithm Most Likely Path (use max instead of sum) G .3 max( .3*.8, .1*.1) *.3 = .072 max( .072*.8, .009*.1) *.3 = .0173 max( .0173*.8, .0014*.1) *.2 =.0028 max( .0028*.8, .0014*.1) *.2 =.00044 G .1 max( .3*.2, .1*.9) *.1 =.009 max( .072*.2, .009*.9) *.1 =.0014 max( .0173*.2+. 0014*.9) *.4 =.0014 max( .0028*.2, .0014*.9 )*.4 =.0005 G C G 0.1 .1 .1 .4 .4 0.9 Non-CpG A A Adapted from David Pollock’s (note error in formulas on his) CpG G C A T 0.8 .3 .3 .2 .2 0.2 G C A T The Viterbi Algorithm Most Likely Path: Backtracking G .3 0.1 .1 .1 .4 .4 0.9 G .1 G max( max( .3*.8, .072*.8, .1*.1) *.3 = .072 .009*.1) *.3 = .0173 max( .3*.2, .1*.9) *.1 =.009 max( .072*.2, .009*.9) *.1 =.0014 C G max( .0173*.8, .0014*.1) *.2 =.0028 max( .0028*.8, .0014*.1) *.2 =.00044 max( max( .0028*.2, .0014*.9 )*.4 =.0005 .0173*.2 +.0014*.9) *.4 =.0014 A A Adapted from David Pollock’s Non-CpG CpG G C A T 0.8 .3 .3 .2 .2 0.2 G C A T Forward-backward algorithm G .3 0.1 .1 .1 .4 .4 0.9 Non-CpG G .1 (.3*.8+ .1*.1) *.3 =.075 (.075*.8+ (.3*.2+ .1*.9) *.1 =.015 (.0185*.8 (.003*.8+ +.0029*.1 .0025*.1) *.3 =.0185 )*.2 =.003 *.2 =.0005 (.075*.2+ .015*.9) *.1 =.0029 (.0185*.2 (.003*.2+| .0025*.9) *.4 =.0011 .015*.1) +.0029*.9) *.4 =.0025 Problem 3: How G C to learnGmodel? A A Forward algorithm calculated Pr(O1..t,Xt=i| λ) Parameter estimation by Baum-Welch Forward Backward Algorithm Forward variable αt(i) =Pr(O1..t,Xt=i | λ) Backward variable βt(i) =Pr(Ot+1..N|Xt=i, λ) Rabiner 1989 Homology HMM • Gene recognition, classify to identify distant homologs • Common Ancestral Sequence – – – – – Parameter set λ = (A, B, π), strict left-right model Specially defined set of states: start, stop, match, insert, delete For initial state distribution π, use ‘start’ state For transition matrix A use global transition probabilities For emission matrix B • Match, site-specific emission probabilities • Insert (relative to ancestor), global emission probs • Delete, emit nothing • Multiple Sequence Alignments Adapted from David Pollock’s Homology HMM insert start insert insert match delete match delete end Adapted from David Pollock’s Homology HMM Example A C D E F .1 .05 .2 .08 .01 match A C D E F .04 .1 .01 .2 .02 match A C D E F .2 .01 .05 match .1 .06 Ungapped blocks Ungapped blocks where insertion states model intervening sequence between blocks Insert/delete states allowed anywhere Allow multiple domains, sequence fragments Eddy, 1998 Center for Genes, Environment, and Health 24 Homology HMM • Uses – Find homologs to profile HMM in database • Score sequences for match to HMM – Not always Pr(O| λ) since some areas may highly diverge – Sometimes use ‘highest scoring subsequence’ • Goal is to find homologs in database – Classify sequence using library of profile HMMs • Compare alternative models – Alignment of additional sequences – Structural alignment when alphabet is secondary structure symbols so can do fold-recognition, etc Adapted from David Pollock’s Why Hidden Markov Models for MSA? • Multiple sequence alignment as consensus – May have substitutions, not all AA are equal FOS_RAT FOS_MOUSE IPTVTAISTSPDLQWLVQPTLVSSVAPSQTRAPHPYGLPTPSTGAYARAGVV 112 IPTVTAISTSPDLQWLVQPTLVSSVAPSQTRAPHPYGLPTQSAGAYARAGMV 112 – Could use regular expressions but how to handle indels? FOS_RAT FOS_MOUSE FOS_CHICK IPTVTAISTSPDLQWLVQPTLVSSVAPSQTRAPHPYGLPTPS-TGAYARAGVV 112 IPTVTAISTSPDLQWLVQPTLVSSVAPSQTRAPHPYGLPTQS-AGAYARAGMV 112 VPTVTAISTSPDLQWLVQPTLISSVAPSQNRG-HPYGVPAPAPPAAYSRPAVL 112 – What about variable-length members of family? FOS_RAT FOS_MOUSE FOS_CHICK FOSB_MOUSE FOSB_HUMAN IPTVTAISTSPDLQWLVQPTLVSSVAPSQ-------TRAPHPYGLPTPS-TGAYARAGVV IPTVTAISTSPDLQWLVQPTLVSSVAPSQ-------TRAPHPYGLPTQS-AGAYARAGMV VPTVTAISTSPDLQWLVQPTLISSVAPSQ-------NRG-HPYGVPAPAPPAAYSRPAVL VPTVTAITTSQDLQWLVQPTLISSMAQSQGQPLASQPPAVDPYDMPGTS----YSTPGLS VPTVTAITTSQDLQWLVQPTLISSMAQSQGQPLASQPPVVDPYDMPGTS----YSTPGMS Center for Genes, Environment, and Health 112 112 112 110 110 26 Why Hidden Markov Models? • Rather than consensus sequence which describes the most common amino acid per position, HMMs allow more than one amino acid to appear at each position • Rather than profiles as position specific scoring matrices (PSSM) which assign a probability to each amino acid in each position of the domain and slide fixed-length profile along a longer sequence to calculate score, HMMs model probability of variable length sequences • Rather than regular expressions which can capture variable length sequences yet specify a limited subset of amino acids per position, HMMs quantify difference among using different amino acids at each position Center for Genes, Environment, and Health 27 Model Comparison • Based on P(D | , M) – For ML, take Pmax (D | , M) • Usually ln Pmax (D | , M) – For heuristics, “score” is – For Bayesian, calculate to avoid numeric error log 2 P(D | fixed , M) P(D | , M) * P * P M Pmax (, M | D) P(D | , M) * P * P M – Uses ‘prior’ information on parameters P ( ) Adapted from David Pollock’s Parameters, • Types of parameters – – – – – – Amino acid distributions for positions (match states) Global AA distributions for insert states Order of match states Transition probabilities Phylogenetic tree topology and branch lengths Hidden states (integrate or augment) • Wander parameter space (search) – Maximize, or move according to posterior probability (Bayes) Adapted from David Pollock’s Expectation Maximization (EM) • Classic algorithm to fit probabilistic model parameters with unobservable states • Two Stages – Maximize • If know hidden variables (states), maximize model parameters with respect to that knowledge – Expectation • If know model parameters, find expected values of the hidden variables (states) • Works well even with e.g., Bayesian to find near-equilibrium space Adapted from David Pollock’s Homology HMM EM • Start with heuristic MSA (e.g., ClustalW) • Maximize – Match states are residues aligned in most sequences – Amino acid frequencies observed in columns • Expectation – Realign all the sequences given model • Repeat until convergence • Problems: Local, not global optimization – Use procedures to check how it worked Adapted from David Pollock’s Model Comparison • Determining significance depends on comparing two models (family vs non-family) – Usually null model, H0, and test model, H1 – Models are nested if H0 is a subset of H1 – If not nested • Akaike Information Criterion (AIC) [similar to empirical Bayes] or • Bayes Factor (BF) [but be careful] • Generating a null distribution of statistic – Z-factor, bootstrapping, , parametric bootstrapping, posterior predictive 2 Adapted from David Pollock’s Z Test Method • Database of known negative controls – E.g., non-homologous (NH) sequences ~ N(, ) – Assume NH scores • i.e., you are modeling known NH sequence scores as a normal distribution – Set appropriate significance level for multiple comparisons (more below) • Problems – Is homology certain? – Is it the appropriate null model? • Normal distribution often not a good approximation – Parameter control hard: e.g., length distribution Adapted from David Pollock’s Bootstrapping and Parametric Models • Random sequence sampled from the same set of emission probability distributions – Same length is easy – Bootstrapping is re-sampling columns – Parametric uses estimated frequencies, may include variance, tree, etc. • More flexible, can have more complex null • Pseudocounts of global frequencies if data limit • Insertions relatively hard to model – What frequencies for insert states? Global? Adapted from David Pollock’s Homology HMM Resources • UCSC (Haussler) – SAM: align, secondary structure predictions, HMM parameters, etc. • WUSTL/Janelia (Eddy) – Pfam: database of pre-computed HMM alignments for various proteins – HMMer: program for building HMMs Adapted from David Pollock’s Center for Genes, Environment, and Health 36 Why Hidden Markov Models? • Multiple sequence alignment as consensus – May have substitutions, not all AA are equal FOS_RAT FOS_MOUSE IPTVTAISTSPDLQWLVQPTLVSSVAPSQTRAPHPYGLPTPSTGAYARAGVV 112 IPTVTAISTSPDLQWLVQPTLVSSVAPSQTRAPHPYGLPTQSAGAYARAGMV 112 – Could use regular expressions but how to handle indels? FOS_RAT FOS_MOUSE FOS_CHICK IPTVTAISTSPDLQWLVQPTLVSSVAPSQTRAPHPYGLPTPS-TGAYARAGVV 112 IPTVTAISTSPDLQWLVQPTLVSSVAPSQTRAPHPYGLPTQS-AGAYARAGMV 112 VPTVTAISTSPDLQWLVQPTLISSVAPSQNRG-HPYGVPAPAPPAAYSRPAVL 112 – What about variable-length members of family? FOS_RAT FOS_MOUSE FOS_CHICK FOSB_MOUSE FOSB_HUMAN IPTVTAISTSPDLQWLVQPTLVSSVAPSQ-------TRAPHPYGLPTPS-TGAYARAGVV IPTVTAISTSPDLQWLVQPTLVSSVAPSQ-------TRAPHPYGLPTQS-AGAYARAGMV VPTVTAISTSPDLQWLVQPTLISSVAPSQ-------NRG-HPYGVPAPAPPAAYSRPAVL VPTVTAITTSQDLQWLVQPTLISSMAQSQGQPLASQPPAVDPYDMPGTS----YSTPGLS VPTVTAITTSQDLQWLVQPTLISSMAQSQGQPLASQPPVVDPYDMPGTS----YSTPGMS 112 112 112 110 110 – (but don’t accept everything – typically introduce gap penalty) Center for Genes, Environment, and Health 37 Why Hidden Markov Models? • Rather than consensus sequence which describes the most common amino acid per position, HMMs allow more than one amino acid to appear at each position • Rather than profiles as position specific scoring matrices (PSSM) which assign a probability to each amino acid in each position of the domain and slide fixed-length profile along a longer sequence to calculate score, HMMs model probability of variable length sequences • Rather than regular expressions which can capture variable length sequences yet specify a limited subset of amino acids per position, HMMs quantify difference among using different amino acids at each position Center for Genes, Environment, and Health 38 Acknowledgements Center for Genes, Environment, and Health 39