Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Polysubstance dependence wikipedia , lookup
Psychopharmacology wikipedia , lookup
Neuropsychopharmacology wikipedia , lookup
Orphan drug wikipedia , lookup
Neuropharmacology wikipedia , lookup
Compounding wikipedia , lookup
Theralizumab wikipedia , lookup
Pharmacognosy wikipedia , lookup
Pharmacogenomics wikipedia , lookup
Drug design wikipedia , lookup
Drug interaction wikipedia , lookup
Pharmaceutical industry wikipedia , lookup
Drug discovery wikipedia , lookup
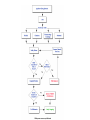
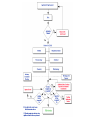
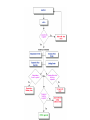
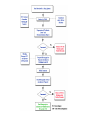
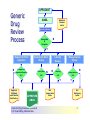
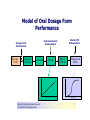
Over-the-Counter Drug Products Over-the-Counter (OTC) drug products are those drugs that are available to consumers without a prescription. There are more than 80 classes (therapeutic categories) of OTC drugs, ranging from acne drug products to weight control drug products. As with prescription drugs, CDER oversees OTC drugs to ensure that they are properly labeled and that their benefits outweigh their risks. OTC drugs play an increasingly vital role in America's health care system by providing easy access to certain drugs that can be used safely without the help of a health care practitioner. This enables consumers to take control of their own health care in many situations. There are more than 100,000 OTC drug products marketed, encompassing about 800 significant active ingredients. Most OTC drug products have been marketed for many years, prior to the laws that require proof of safety and effectiveness before marketing. For this reason, FDA has been evaluating the ingredients and labeling of these products as part of "The OTC Drug Review Program." The goal of this program is to establish OTC drug monographs for each class of products. OTC drug monographs are a kind of "recipe book" covering acceptable ingredients, doses, formulations, and labeling. Monographs will continually be updated adding additional ingredients and labeling as needed. Products conforming to a monograph may be marketed without further FDA clearance, while those that do not, must undergo separate review and approval through the "New Drug Approval System." The NDA system--and not the monograph system--is also used for new ingredients entering the OTC marketplace for the first time. For example, the newer OTC products [previously available only by prescription] are first approved through the NDA system and their "switch" to OTC status is approved via the NDA system. FDA's review of OTC drugs is primarily handled by CDER's Division of Over-the-Counter Drug Products in the Office of Drug Evaluation V. However, scientists and regulators throughout CDER, the Office of General Counsel, and other Centers within FDA are routinely asked to assist in this massive effort. There is also an advisory committee, "The Nonprescription Drug Advisory Committee," which meets regularly to assist the agency in evaluating issues surrounding these products. The FDA Process for Approving Generic Drugs Gary J. Buehler, R.Ph. Dale Conner, Pharm. D. Director Director, Division of Bioequivalence Office of Generic Drugs Did you know that generic drugs... • Are safe and effective alternatives to brand name prescriptions • Can help both consumers and the government reduce the cost of prescription drugs • Are currently used in 50% of all prescriptions dispensed • Save an average of $50 for every prescription sold Center for Drug Evaluation & Research U.S. Food & Drug Administration 2 Hatch-Waxman Amendments to FFD&C Act - 1984 • Considered one of the most successful pieces of legislation ever passed • Created the generic drug industry • Increased availability of generics • 1984 12% prescriptions were generic • 2000 44% prescriptions were generic - yet only 8% of revenue for prescription drugs • Compromise legislation to benefit both brand and generic firms Continued Center for Drug Evaluation & Research U.S. Food & Drug Administration 3 Hatch-Waxman Amendments to FFD&C Act - 1984 • Allowed generic firms to rely on findings of safety and efficacy of innovator drug after expiration of patents and exclusivities (do not have to repeat expensive clinical and pre-clinical trials) • Allowed patent extensions and exclusivities to innovator firms Center for Drug Evaluation & Research U.S. Food & Drug Administration 4 NDA vs. ANDA Review Process Brand Name Drug NDA Requirements Generic Drug ANDA Requirements 1. 2. 3. 4. 5. 6. 7. 8. 1. 2. 3. 4. 5. Chemistry Manufacturing Controls Labeling Testing Animal Studies Clinical Studies Bioavailability Center for Drug Evaluation & Research U.S. Food & Drug Administration Chemistry Manufacturing Controls Labeling Testing 6. Bioequivalence 5 What are the requirements for a generic drug? • Labeling • Chemistry/Microbiology • Bioequivalence • Legal Center for Drug Evaluation & Research U.S. Food & Drug Administration 6 How do we assure the quality of generic drugs? • First 5 steps of review process are identical to NDA process • Bioequivalence for complicated products is discussed with the same staff that reviewed the brand product • FDA has experience with the product • Scientific literature published • Product is known to be safe Center for Drug Evaluation & Research U.S. Food & Drug Administration 7 APPLICANT Generic Drug Review Process ANDA Refuse to Receive Letter Application Review Acceptable & Complete N Y Request for Plant Inspection N Chemistry & Micro Review PreApproval Inspection Results OK? Chem/Micro N OK? Y Approval Withheld until Results Satisfactory N Labeling Review APPROVED ANDA Center for Drug Evaluation & Research U.S. Food & Drug Administration Labeling Bioequivalence OK? OK? Y Y Not Approvable Letter Bioequivalence Review N Y Bio Deficiency Letter 8 What are the requirements for a generic drug? • Same active ingredient(s) • Same route of administration • Same dosage form • Same strength • Same conditions of use Compared to reference listed drug (RLD) - (brand name product) Center for Drug Evaluation & Research U.S. Food & Drug Administration 9 Labeling • “Same” as brand name labeling • May delete portions of labeling protected by patent or exclusivity • May differ in excipients, PK data and how supplied Center for Drug Evaluation & Research U.S. Food & Drug Administration 10 Chemistry • Components and composition • Manufacturing and controls • Batch formulation and records • Description of facilities • Specs and tests • Packaging • Stability Center for Drug Evaluation & Research U.S. Food & Drug Administration 11 Manufacturing Compliance Programs • Purpose - To assure quality of marketed drug products • Mechanisms - Product Testing – Surveillance – Manufacturing/Testing plant inspections – Assess firm’s compliance with good manufacturing processes Center for Drug Evaluation & Research U.S. Food & Drug Administration 12 APPROVED DRUG PRODUCTS WITH THERAPEUTIC EQUIVALENCE EVALUATIONS 23rd EDITION THE PRODUCTS IN THIS LIST HAVE BEEN APPROVED UNDER SECTION 505 OF THE FEDERAL FOOD, DRUG, AND COSMETIC ACT. U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES PUBLIC HEALTH SERVICE FOOD AND DRUG ADMINISTRATION CENTER FOR DRUG EVALUATION AND RESEARCH OFFICE OF GENERIC DRUGS 2003 Electronic Orange Book - http://www.fda.gov/cder/ob/ Center for Drug Evaluation & Research U.S. Food & Drug Administration 13 “Orange Book” Book • All FDA approved drug products listed (NDA’s, OTC’s & ANDA’s) – Therapeutic equivalence codes “A” = Substitutable “B” = Inequivalent, NOT Substitutable – Expiration dates: patent and exclusivity – Reference Listed Drugs/brand drugs identified by FDA for generic companies to compare with their proposed products Center for Drug Evaluation & Research U.S. Food & Drug Administration 14 Definition of Bioequivalence Pharmaceutical equivalents whose rate and extent of absorption are not statistically different when administered to patients or subjects at the same molar dose under similar experimental conditions Center for Drug Evaluation & Research U.S. Food & Drug Administration 15 Purpose of BE • Therapeutic equivalence (TE) • Bioequivalent products can be substituted for each other without any adjustment in dose or other additional therapeutic monitoring • The most efficient method of assuring TE is to assure that the formulations perform in an equivalent manner Center for Drug Evaluation & Research U.S. Food & Drug Administration 16 Model of Oral Dosage Form Performance Dosage Form Drug in Solution Clinical/PD Measurement Pharmacokinetic Measurement Dosage Form Performance Gut Wall Blood Dose Center for Drug Evaluation & Research U.S. Food & Drug Administration Site of Activity Therapeutic Effect ln Dose 17 Clinical/PD Response Clinical/PD Dose-Response Log Dose Center for Drug Evaluation & Research U.S. Food & Drug Administration 18 Plasma Conc. Plasma Concentration-Dose Dose Center for Drug Evaluation & Research U.S. Food & Drug Administration 19 Approaches to Determining Bioequivalence (21 CFR 320.24) • In vivo measurement of active moiety or moieties in biologic fluid FeV Albuterol Blanching Study • In vivo pharmacodynamic Topical Corticosteroid comparison Topicals • In vivo limited clinical Nasal Suspensions comparison Questran - Binding Studies • In vitro comparison Nasal Solutions-Sprayer 1 • Any other approach deemed appropriate by FDA Center for Drug Evaluation & Research U.S. Food & Drug Administration Evaluation Propofol - Droplet Size 20 Study Designs • Single-dose, two-way crossover, fasted • Single-dose, two-way crossover, fed • Alternatives – Single-dose, parallel, fasted – Single-dose, replicate design – Multiple-dose, two-way crossover, fasted – Clinical endpoint study Center for Drug Evaluation & Research U.S. Food & Drug Administration Long Half-Life (wash-out) Amiodarone, Etidronate Highly Variable Drugs Less Sensitive Clozapine (Patient Trials) Chemotherapy Trials Topicals Nasal Suspensions 21 Waivers of In Vivo Study Requirements • Definition • Criteria (21 CFR 320.22) – In vivo bioequivalence is self-evident – Parenteral solutions – Inhalational anesthetics – Topical (skin) solution – Oral solution – Different proportional strength of product with demonstrated BE Center for Drug Evaluation & Research U.S. Food & Drug Administration 22 Statistical Analysis (Two One-sided Tests Procedure) • AUC and Cmax – 90% Confidence Intervals (CI) must fit between 80%-125% Center for Drug Evaluation & Research U.S. Food & Drug Administration 23 Statistical Analysis 80 - 125 % • What does this mean? • Can there be a 46% difference? • What is a point estimate? • What is a confidence interval? Center for Drug Evaluation & Research U.S. Food & Drug Administration 24 Statistical Analysis • Bioequivalence criteria – Two one-sided tests procedure • Test (T) is not significantly less than reference • Reference (R) is not significantly less than test • Significant difference is 20% ( = 0.05 significance level) – T/R = 80/100 = 80% – R/T = 80% (all data expressed as T/R so this becomes 100/80 = 125%) Center for Drug Evaluation & Research U.S. Food & Drug Administration 25 Possible BE Results (90% CI) 80% Center for Drug Evaluation & Research U.S. Food & Drug Administration T/R (%) 125% 26 Narrow Therapeutic Range (NTI) Drugs • Drug Products that are subject to therapeutic drug concentration or pharmacodynamic monitoring – Examples are: Digoxin, Lithium, Phenytoin, Warfarin • Traditional bioequivalence limit of 80125% is unchanged for these products Center for Drug Evaluation & Research U.S. Food & Drug Administration 27 Contacting the OGD Contact: Office of Generic Drugs FDA/CDER (HFD-600) 7500 Standish Place Rockville, MD 20855 phone: 301-827-5845 Web site: www.fda.gov/cder/ogd/index.htm Center for Drug Evaluation & Research U.S. Food & Drug Administration 28