Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
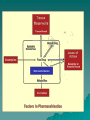
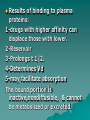
PHARMACOKINETICS Definition: quantitative study of drug absorption, distribution, metabolism, and excretion (ADME), and their mathematical relationship. Disposition: term used to describe the simultaneous effect of the distribution and elimination (processes subsequent to absorption). Absorption Absorption: passage of drug through body barriers. Transmembrane movement of drugs: Passive transfer: – Simple diffusion – Filtration N.B.: Diffusion is most important means. It is affected by: size of molecule, conc. Gradient, surface area, permeability coefficient. (FICK'S Law). N.B.: Permeability coeffiecient varies according to the concentration of ionized and nonionized form of the drug on both sides of the membrane, determined by pka of the drug. Henderson Hasselbalch equation pH=pka + log [nonprotonated species/ protonated species] Specialized transport – Active transport 1-1ry active transport 2- 2ndry active transport (Cotransport,countertransport) – Facilitated diffusion – Pinocytosis and phagocytosis Bioavailability: percentage of drug released from a formulation that becomes available for biological effect. Factors affecting bioavailability: a)Factors affecting absorption from GIT: 1-Drug 2-Formulation 3-Patients 4-Food. b) First pass effect: presystemic elimination.It is the metabolism of some drugs in a single passage through the liver, gut walls, or lungs before reaching the systemic circulation. Hepatic: e.g.:propranolol, nitroglycerin,cortisone. Pulmonary: isoprenaline,nicotine. Intestinal: tyramine,morphine. *Pulmonary Absorption *Absorption from subcutaneous tissues *Methods for delaying absorption: a.oral b.parenteral. Distribution Passage of drug between blood and tissues. Compartment concept: - one-compartment model -Two- compartment model Mathematically, apparent volume of distribution (Vd): apparent volume that would accommodate all drug in the body if its conc. throughout the body was the same as that in the plasma (not real). N.B.: small volume of distribution is favored by low lipid solubility, high degree of plasma protein binding and low level of tissue binding, & vice-versa. Vd = amount of drug in the body / plasma concentration. Importance: -extent of tissue up-take -calculation of loading dose - dialysis Factors affecting distribution of drugs: 1-Physicochemical properties 2-Size of the tissue and its blood flow. 3-Binding to plasma proteins: a- Albumin b-α-1-acid glycoprotein 4-Binding to cell and tissue components Results of binding to plasma proteins: 1-drugs with higher affinity can displace those with lower. 2-Reservoir 3-Prolongs t 1/2. 4-Determines Vd 5-may facilitate absorption The bound portion is inactive,nondiffusible, & cannot be metabolized or excreted. –BBB –Placental barrier Biotransformation"metabolism" Def.: changes that occur to drug after absorption until excretion. Consequences: -abolishes activity -promotes activity -changes active to another active form. Types of reaction: -Non-synthetic (phase-I): oxidation, reduction, and hydrolysis. -Synthetic (phase-II): adds a constituent to the drug to form highly polar rapidly eliminated conjugates"Conjugation". These include: glucoronic acid,Nacetylation,glycine and sulphuric acid conjugation. Factors affecting biotransformation: species,age,sex, pathological conditions,environmental factors. Microsomal enzymes: SER of liver. 1-MFO, that requires cytochrome P-450, NADPH,molecular oxygen 2-metabolize lipid soluble to water soluble drugs 3-metabolize drugs as well s body as body constituents 4-lack substrate specificity 5-not well developed in newborn. Non-microsomal enzyme systems: e.g.peroxidases, oxidases,and dehydrogenases. N.B.: Enzyme induction Enzyme inhibition. Drug Clearance A measure of the body's ability to eliminate drugs. It's defined as the volume of a fluid from which all drug is removed per unit time. CLEARNCE (CL)= Rate of elimination / Drug conc. May involve: kidney, liver, lungs, and other organs. Factors affecting clearance: – Blood flow – binding to plasma proteins – activity of the processes involving blood removal(e.g.: hepatic enzymes…etc). N.B.: hepatic clearance depends on: intrinsic activity of metabolic enzymes and transport processes, free fraction of drug and total effective blood flow. Elimination half-life: the time required to reduce the plasma concentration of drug to 1/2 the initial conc. t (1/2)=0.693 Vd / CL Value of t (1/2): -time required to attain Css. -Determines dosage interval ( usually = t 1/2) -Index of drug elimination Firstorder kinetics: (non-saturable) Clearance is directly proportional to the conc. o the drug in the plasma, i.e.; the rate of elimination is exponentially related to the amount of drug available. This means that a constant fraction or ratio of drug is eliminated per unit time. Characteristics: Constant t1/2. AUC, Css and amount of drug excreted unchanged in urine; all are proportional to the dose. Composition of the drug metabolites excreted is independent of the dose. Zero-order kinetics (non-linear): Drug elimination occurs at a constant rate of the amount of drug to be eliminated. This means that a constant amount of drug being eliminated per unit time. Characteristics: 1. Half-life increases with dose 2. AUC, Css and amount of drug excreted unchanged in urine; all not proportional to the dose. 3. Composition of the drug metabolites excreted may vary with the dose. Saturable kinetics: The elimination does not follow first-order kinetics throughout the dose range. Clearance will vary depending on the achieved drug concentration. Many ADME processes occur via enzymes or carriermediated systems which for these drugs become saturated in the therapeutic range and results in saturable non-linear elimination kinetics. Modest changes in dose or bioavailaility of these drugs may produce unexpected toxicity.