Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Bioequivalence
Accomplishments, Ongoing
Initiatives, and Remaining
Challenges
Marilyn N. Martinez, Ph.D.
US Food and Drug Administration
Center for Veterinary Medicine
Presentation Outline
• Initiatives, past and present
• Challenges
–
–
–
–
Biosimilars
Highly variable drugs
Long T1/2 drugs
Biowaivers
• Scientific “thoughts to consider”
– Why we should avoid interspecies extrapolation of BE
assessments
– Food effects
• Summary of remaining issues
INITIATIVES
Past and Present
Accomplishment:
2010 AAVPT/ECVCP/EAVPT BE
Initiative
EAVPT
2009
A Journey Through
Bioequivalence
Marilyn N. Martinez, Ph.D.
US Food and Drug Administration
Center for Veterinary Medicine
The Plan
EAVPT
2009
Marilyn Martinez and Robert Hunter will publish a stimuli articl e
describing some of the bioequivalence challenges we face in
veterinary medicine.
Prior to the workshop, a webinar will be convened to review basi c
bioequivalence concepts.
The workshop will provide an opportunity to discuss the various
critical challenges facing the evaluation of bioequivalence in
veterinary medicine.
A workshop summary will be prepared and the manuscript
published.
The workshop will serve as a springboard for at least 10 working
groups that will be tasked with developing white papers on the
various complex issues discussed during the workshop. The white
papers are intended to lead to:
Points of concurrence, based upon existing information.
The identification of points necessitating additional discussions.
discussions.
Identification of areas needing additional research.
Current Initiative:
Facing a Global Marketplace
Effort to Foster International
Harmonization
The Veterinary International Conference
on Harmonization (VICH) is in the process
of developing a blood level BE.
Even with the anticipated successful
completion of this blood level BE guideline,
there remain numerous challenges facing
international harmonization –
A potential future VICH initiative?
Numerous differences exist in bioequivalence policies across these
international guidelines (see summary of these differences as written
by Dr. Chantal Lainesse IntegRxal Consulting Strategies
Saskatoon, Saskatchewan CANADA) in an upcoming issue of the
AAPS Journal
Issues Outside the Scope of the VICH
BE Guideline Include:
There also are numerous challenges
facing the global scientific community in
identifying potential solutions to challenges
for complex bioequivalence situations.
Challenges
Biosimilars
• Unlike most prescription drugs made through chemical
processes, biological products are generally made from
human and/or animal materials.
• What defines whether or not two biological products are
similar? Within the U.S., Section 351(i) of the PHS Act
defines biosimilarity to mean that “the <me-too> biological
product is highly similar to the reference product
notwithstanding minor differences in clinically inactive
components” and that “there are no clinically meaningful
differences between the biological product and the
reference product in terms of the safety, purity, and
potency of the product.”
http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm291232.htm
Biosimilars
In Feb, 2012, the FDA the Center for (human)
Drug Evaluation and Research (CDER) and the
Center for (human) Biologics Evaluation and
Research (CBER) has issued three draft
guidances on the assessment of “biosimilar”
products:
– Scientific Considerations in Demonstrating
Biosimilarity to a Reference Product
– Quality Considerations in Demonstrating Biosimilarity
to a Reference Protein Product
– Biosimilars: Questions and Answers Regarding
Implementation of the Biologics Price Competition
and Innovation Act of 2009
http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm291232.htm
Biosimilars
Scientific Considerations in Demonstrating Biosimilarity to a
Reference Product
– This draft guidance describes a risk-based “totality-of-theevidence” approach to evaluate the data and information
submitted in support of a determination of biosimilarity of
the proposed product to the reference product.
– Biosimilar products will be the subject of a New Drug
Application (NDA) submitted under section 505(b)(2) of the
Federal Food, Drug, and Cosmetic Act (FD&C Act).
– Comparability to the Listed Reference Product can include
a comparison with respect to structure, function, animal
toxicity, human pharmacokinetics (PK) and
pharmacodynamics (PD), clinical immunogenicity, and
clinical safety and effectiveness.
http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/
Guidances/UCM291128.pdf
Biosimilars
Under section 351(k) of the PHS Act, the application must
contain, among other things, information demonstrating that “the
biological product is biosimilar to a reference product” based
upon data derived from:
»Analytical studies that demonstrate that the biological product
is highly similar to the reference product notwithstanding minor
differences in clinically inactive components;
»Animal studies (including the assessment of toxicity); and
»A clinical study or studies (including the assessment of
immunogenicity, and PK or PD) that are sufficient to demonstrate
safety, purity, and potency in one or more appropriate conditions
of use for which the reference product is licensed and intended to
be used and for which licensure is sought
The Agency has the discretion to determine that an element
described above is unnecessary.
.
http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/
Guidances/UCM291128.pdf
A Potential Hurdle in the Approval of
“Biosimilars” for Veterinary Use
•Within the human drug regulations, Section 505(b)(2)
allows for an New Drug Application (NDA) that contains full
reports of investigations of safety and effectiveness, where
at least some of the information required for approval
comes from studies not conducted by or for the
applicant and for which the applicant has not obtained
a right of reference (i.e., - generic-like application) or use.
• Any aspects of the proposed product that differ from the
listed drug must be supported by adequate data and
information to show that the differences do not affect the
safety and effectiveness of the proposed product.
There currently are no similar US regulations that
cover “505(b)(2)-like applications” for veterinary drugs.
Three Tough Issues from
2010 BE Workshop
• How to handle situations when the drug is
systemically absorbed but where the blood
levels are highly variable?
• How to evaluate bioequivalence for drugs
and/or dosage forms with very long T½’s?
• Can we expand the criteria for granting
biowaivers while maintaining confidence in
our BE determinations?
Issue 1: Highly Variable Drugs
• When extended crossover (i.e., 3 or 4-periods) designs
can be employed, we can use a scaled average
bioequivalence (SABE) approach (see Claxton et al.,
JVPT, 35(suppl 1) 11-16, 2012) where the confidence
bounds are adjusted by the within-subject variation of the
reference treatment.
• When the study necessitates the use of a parallel study
design, the situation if far more complicated and there
are no existing paradigms that adequately address this
situation. Potential solutions:
– Can population approaches (nonlinear mixed effect models) be
used to demonstrate comparability through a comparison of
population predicts rather than by traditional confidence interval
approaches?
– Could we use alternative designs that allow for between-subject
but within-treatment estimates of variability to permit some form
of SABE approach?
Issue 2: Evaluation of Long T½
Drugs and Drug Products
• For slow depleting drugs in immediate
release formulations, once the drug is
completely absorbed, there is a duration of
sampling that will maximize the precision
of the comparative bioavailability
comparison (minimizing the impact of
noise derived from measuring low drug
concentrations or variability due to small
differences in elimination rates).
Gehring and Martinez, 2012, JVPT, 35(Suppl 1):3-9.
Issue 2: Evaluation of Long T½
Drugs and Drug Products
• For extended release products, there are
concerns that may extend beyond simply AUC
and Cmax comparisons:
– When Tmax occurs may be critical to product
performance.
– There may be multiple maxima.
– The duration of exposure may be an important issue.
• As discussed in the paper, it will be important to
identify the shape of the innovator product and
its relationship to effect when assessing
appropriate blood level BE criteria for these
dosage forms.
Gehring and Martinez, 2012, JVPT, 35(Suppl 1):3-9.
Issue 2: Evaluation of Long T½ Drugs and
Drug Products
For extended release products,
there may be a need to estimate
partial AUC’s.
In this example, two products have
equivalent AUC0-tlast and Cmax, but
differing shapes. These differences
are clinically relevant.
Endrenyi and
Tothfalusi, 2010, J.
Pharm Pharmaceut
Sci, 13:107-113,
Issue 3: Exploring that Criteria for
Granting Biowaivers
Applying a “totality of evidence” approach to identify the
vaiables that will influence product absorption
characteristics.
Can we begin to utilize the Quality by Design (QbD)
concept to support biowaivers?
See Yu, 2008, Pharm Res, 25:781-791.
ICH Q8R2 dated 2009
http://www.fda.gov/downloads/AdvisoryCommittees/Commi
tteesMeetingMaterials/Drugs/AdvisoryCommitteeforPharma
ceuticalScienceandClinicalPharmacology/UCM179424.pdf
CDER: Biowaivers May be Granted for Some
Topical (non-systemcially Absorbed) Products
• Q1: Qualitative Similarity
– Same components
L Yu, 2009
• Q2: Quantitative Similarity
– Same amounts of the same components
• Q3: Structural Similarity
– Same amounts of the same components
arranged in the same way
How do you measure Q3?
What does Q3 similarity imply about bioequivalence?
Definition of Q3
• Structural Similarity
– Arrangement of
matter
– State of aggregation
From Yu,
2009
Example: Polymorphs
Form A
Form B
Same Substance
Q3 is different?
Example: Suspension
A
B
Q: How does
one statistically
compare
particle size?
Same Substance
Q3 is different?
Q1 and Q2 is Not Sufficient:
Why Q3 is Necessary
These kinds of
excipient variations
can impact in vivo
and in vitro product
performance. The
impact of materials
variability is the
subject of ongoing
research.
Patrick Noonan, 2009 CDER Advisory Committee meeting: BE recommendations
for {locally acting} vancomycin HCl products
http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/D
rugs/AdvisoryCommitteeforPharmaceuticalScienceandClinicalPharmacology/UCM1
79424.pdf
Issue 3: Biowaivers, Q1, Q2, Q3 and the
Importance of Process Understanding
• What is Quality by Design (QbD)? QbD has been defined
as “a systematic approach to development that begins with
predefined objectives and emphasizes product and process
understanding and process control, based on sound
science and quality risk management” (ICH Q8).
• QbD is based upon the concept that quality should be built
into the product. It is founded upon understanding the
relationship between the drug, the excipients, and the
manufacturing process. Through this understanding, the
product design space can be defined.
Issue 3: Biowaivers, Q1, Q2, Q3 and the
Importance of Process Understanding
The design space is the
multidimensional
combination of factors that
Control
Control
have been demonstrated to
Control
Space
Space
Space
provide the desirable
product quality. It represents
an optimized set of
A. The control
B. The control space is
C. The control
space is much
adjacent to the limits of
space falls outside
conditions whereby the
smaller than the
the design space. This
of design space.
design
space.
This
system
IS
NOT
This is FAILED
formulation and process
reflects a ROBUST
ROBUST and therefore
control strategy.
process.
more stringent controls
variables give rise to a
are needed.
product that will have the
The relationship between control
desired in vivo performance
strategy and design space.
in the targeted patient
population.
Fahmy, Danielsonn, Martinez, 2012: in Animal Health Drug
Delivery, MJRathbone, Editor
Point to ponder: Can process understanding
be used to support biowaivers? If so, what
information is needed? How could such an
approach help support the available of safe
and effective veterinary medicines?
-Innovator product range of “waiverable”
changes?
-Generic product via Q1, Q2, Q3 +
dissolution?
Could this type of information be superior
to clinical endpoint BE studies?
Effect of change in pathogen susceptibility on clinical endpoint
decision
Example: if the pathogen
susceptibility changes
over time, products that
appear to be clinically
equivalent at one point in
time may not be clinically
equivalent if a shift in
pathogen susceptibility
occurs. Traditional
Therefore other approaches might
clinical endpoint BE trials
provide more exact product
provide no information on
comparisons.
true differences in the
rate and extent of drug
exposure.
Response (percent success)
100
90
80
Less susceptible
70
wild-type responders
60
50
40
30
20
10
0
0
2
4
6
8
Exposure
10
12
14
Issue 3: Exploring the Criteria for
Granting Biowaivers
• Drug solubility will influence the potential impact
of formulation on in vivo product performance:
the greater the aqueous solubility, the more
robust the drug absorption characteristics
relative to formulation variations.
• For injectable formulations, the low fluid volume
will render it far more difficult to classify a
compound as fully soluble, even if a drug is
administered as a parenteral solution.
Issue 3: Exploring the Criteria for
Granting Biowaivers
• Traditional approach: CDER BCS Biowaiver Guidance:
Biowaiver is based upon the Biopharmaceutics
Classification System (BCS):
– Drugs eligible for a biowaiver need to be classified as highly
soluble and highly permeable (BCS Class 1).
– The product is an rapidly dissolving dosage form intended for
oral administration.
– Rapidly dissolving is defined as no less than 85% of the labeled
amount of the drug substance dissolves within 30 minutes, using
U.S. Pharmacopeia (USP) Apparatus I at 100 rpm (or Apparatus
II at 50 rpm) in a volume of 900 ml or less in each of the
following media: (1) 0.1 N HCl or Simulated Gastric Fluid USP
without enzymes; (2) a pH 4.5 buffer; and (3) a pH 6.8 buffer or
Simulated Intestinal Fluid USP without enzymes.
Difficulty in Applying CDER Approach to
Veterinary Formulations.
• BCS complications:
– We have not established the criteria for defining “highly
soluble” across the various animal species.
– There are no standardized methods for evaluating drug
permeability.
– Many of the drugs for which biowaiver will be needed will
be non-systemically absorbed or locally acting
compounds.
• Dissolution complications:
– There are several product categories that are not
amenable to traditional in vitro dissolution test methods.
– Differences in GI transit time renders it inappropriate to
strive for a “one size fits all” approach to defining a
formulation as being “rapidly dissolving”.
– What pH is appropriate for testing dissolution across the
various animal species?
Difficulty in Applying CDER Approach
to Veterinary Formulations
• During our bioequivalence workshop, there were
some individuals that expressed concerns with the
waiving of medicate premixes solely on the
classification of highly soluble as described by the
CDER Biowaiver Guidance (see Hunter et al., 2012,
JVPT, 35 (Suppl 1):53-63). Conversely, there were
others who questioned whether current definitions
for highly soluble may be too rigid for some animal
species (such as ruminants).
• Therefore, additional work is needed in this area:
– Potentially applying the QbD and Q1, Q2, Q3
concept
– Better definition of drug solubility
New Initiative: US Pharmacoepeia: Defining
highly soluble compounds in Dogs and Cattle
• Stimuli Article Published:
based largely on the three
solubility manuscripts in the
JVPT Supplement.
• Workshop on defining soluble
in cattle and dogs scheduled
in November 7 and 8, 2012
(USP Headquarters, Rockville
MD)
http://www.usp.org/meetingscourses/workshops/solubilitycriteria-veterinary-products
• Criteria will be incorporated
into a USP General Chapter
for Veterinary Therapeutics.
• Following successful
completion in these two
species, other species will be
considered (e.g., feline, swine
and horse).
Two Additional Scientific Thoughts
to Consider
• Under what conditions is there a risk of a
food-by-formulation interaction (thereby
necessitating BE studies be conducted in
both fed and fasted monogastrics)?
• Why can’t BE determinations be
extrapolated across animal species
(including human to dog)?
Food EffectsNeed to Distinguish Between PK and BE Issues
PK Issues
BE Issues
• Impact on drug
absorption due to
enhanced solubilization
with food.
• Impaired drug absorption
due to chelation or
access to intestinal
absorptive membrane.
• Prolonged gastric
residence with food.
• Altered first pass drug
metabolism
• Impact of food on drug
absorption from modified
release formulations.
• Impact of food on tablet
coating
• Impact of food on particle
size-related differences in
drug solubilization.
• Food-by-excipient
interactions
• Food interaction with
certain taste-masking
resins
Food-By-Drug Interaction
Singh, 2005, Drug Development
Research, 65:55-75.
Highly soluble
Poorly soluble
The impact of food on drug bioavailability is directly correlated with the
dose-to-solubility ratio (SR) where solubility was generally estimated in
water (in some cases in acetate buffer at H 5.0) and the ratio was estimated
as dose/aqueous solubility.
Study Conclusions
• Increasing particle surface area significantly increases
drug absorption
• Food enhances drug absorption through an increase in
solubility
• It should be noted that in general, food will increase the
bioavailability of low solubility compounds if the drug is
well absorbed throughout the GI tract, but will reduce drug
absorption if the compound is absorbed only the upper
small intestine. This may be due to:
– Increased volume in GI tract decreases concentration of drug
exposed to absorptive membrane.
– Increased viscosity decreases interaction of drug and absorptive
membrane.
– Bile salt secretion decreases intermicellar “free” drug fraction in
the upper intestine which could lead to a decrease in drug
absorption
– Interaction between food and drug
– Gastric instability of drug
Cilostazol: Food-by-Formulation Interaction:
Particle Size
• Sub-micron particles (wet milled median particle size = 0.26
μm) had substantially higher bioavailability than the larger jet
milled (2.4 μm) or hammer milled particles (median particle
size = 13 μm).
• Food increased bioavailability of jet milled and hammer-milled
products but slightly decreased bioavailability of wet milled.
AUC
fed/fast =
0.76
AUC
fed/fast
= 3.7
AUC
fed/fast =
1.8
Jinno, et al., 2006, J. Cont.
Rel, 111:56-64.
UNDER NO
CONDITION ARE
PRODUCTS
BIOEQUIVALENT!
Wet milled
Jet milled
Hammer milled
Nelfinavir – Food-By-Formulation Interaction
Film Coated Tablets
• Relative bioavailability tested in 4-period crossover
involving 52 healthy male subjects: 2 formulations under
fed or fasted conditions.
• Food increased AUC 13-fold for reference product and
21-fold for test product.
Kaeser et al., 2005, Int J. Clin
Pharmacol Therap, 43:154-162.
Why Can’t We Simply Extrapolate
BE Between Species?
• Oral Formulations:
– Differences in GI physiology
•
•
•
•
Transit time
Fluid composition
Fluid volumes
Agitation (e.g., much higher crushing force in
canine versus human stomach).
• Dietary differences
• Potential differences in absorption site
Human – Canine Differences in Site of
Absorption: Ciprofloxacin IR Tablets
GastroPlus modeling of published data in Humans and Dogs after oral
administration of immediate release tablets
Human
Dog
Why Can’t We Simply Extrapolate
BE Between Species?
• Parenteral Formulations:
– Differences in injection site physiology
• Fluid composition
• Fluid volumes
• Lymphatic and vascular composition of the
injection site
• Muscle movements
• Inflammatory responses
– Differences in injected volumes
For more information, refer to Martinez, 2011, AAPS Journal, 13:632-649.
Another Key Issue: Profile Sensitivity to Absorption
Rate Reflects Differences in Elimination Rate
As ka approaches (or is less than)
kel, ka has a much greater effect on
Cmax and Tmax. When kel<<ka,
changes in the ka value has a
diminished influence on Cmax
ke=0.05 Cmax .25/.15 = 1.16
80
70
ka=0.25
60
ka=0.2
50
40
30
ka=0.15
20
10
0
0
10
20
30
40
50
60
Ke = 0.1 Cmax .25/.15 = 1.22
60
Ke=0.01 Cmax .25/.15 = 1.06
100
50
ka=0.25
40
ka=0.2
30
ka=0.15
80
60
20
40
10
20
ka=0.25
ka=0.2
ka=0.15
0
0
0
10
20
30
40
50
60
0
10
20
30
40
50
60
Summary of Remaining Issues:
• International harmonization
• Scientific:
–
–
–
–
Highly variable drugs
Multiple peaks/very long T½ drugs
Biosimilars
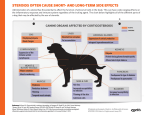
Drugs exhibiting both local and systemic effects
• Regulatory:
– Alternatives to traditional BE studies for nonsystemically absorbed products.
– Methods of evaluating BE for products indicated for
use across multiple animal species.
And the many issues covered in the JVPT 2012 Supplement!