Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
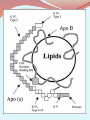
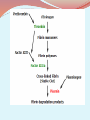
Lipoprotein (a) [Lp(a)] is a heterogeneous lipoprotein that shares many properties with low density lipoprotein (LDL). Plasma Lp(a) consist of a cholestrol-rich LDL particle with one molecule apoprotein B100 and an additional protein, apoprotein (a), attached via a disulfide bond. Elevated Lp(a) levels can potentially increased the risk of CVD. Plasma levels of Lp(a) are nearly similar in men and women, mean values were 14mg/dl for men and 15mg/dl for women with SD of 17 for both sexes. Levels above 30 mg/dl are considered elevated. Lp(a) levels did not significantly differ across the whole age range. METABOLISM It is believed that plasma levels of Lp(a) are determined chiefly by rates of hepatic synthesis of apo(a); although the site of formation of Lp(a) has not been definitively identified, evidence suggests that apo(a) adducts extracellularly and covalently to apo B 100- containing lipoprotein, predominantly LDL. The apo(a) gene has 10 different types of plasminogenlike K4 domains. The first type and type 3-10 repeats are present in only one copy each, while K4 type 2 is present as multiple copies. It is the variable K4 type 2 repeats that are responsible for the heterogeneity in size variation in the apo(a) glycoprotein. Lp(a) size is inversely related to Lp(a) plasma levels. Smaller apo(a) sizes generally correspond to higher plasma levels, but the relationship between Lp(a) isoform size and Lp(a) levels is not fixed. Lp(a) is thought to be catabolized primarily by hepatic and renal pathways, but these metabolic routes do not appear to govern plasma Lp(a) levels. Pathophysiological mechanism underlying the atherothrombotic potential of Lp(a) Lp(a) may bind more avidly retained than LDL to arterial intima as it binds to the extracellullar matrix not only through apo(a), but also by its apo B component, thereby contributing cholesterol to the expanding atherosclerotic plague. Lp(a) binds to several extracellular matrix proteins including fibrin and defensins that are released by neutrophils during inflammation. It is likely that defensins provides a bridge between Lp(a) and the extracellular matrix. Through its apo(a) moiety also interacts with B2integrin, thereby promoting the adhesion of monocytes. Lp(a) has also been shown to bind pro inflammatory oxidized phospholipids. Apo(a), a homologue of fibrinolytic proenzyme plasminogen can competitively inhibit tissue plasminogen activator that mediate plasminogen activation on fibrin surface. Small apa(a) isoform possess high potency of antifibrinolytic effects. * Lp(a) elevations have been linked to increased risk for CHD, ischemic stroke and peripheral vascular disease. In fact, an increase in Lp(a) level is the most common inherited lipid disorder in subjects with premature CHD. Whom to screen for Lp(a) Lp(a) should be measured once in all subjects at intermediate or high risk of CVD/CHD who present with i. Premature CVD ii. Familial hypercholesterolemia iii. A family history of premature CVD/or elevated Lp(a) iv. Recurrent CVD despite statin treatment v. ≥ 3% 10 –year risk of fatal CVD according to the European guidelines and vi. ≥ 10% 10 –year risk of fatal and/or nonfatal CHD according to US guidelines *It is consistently reported that lifestyle modifications such as diet, weight loss, and exercise have little or no effect on Lp(a) levels . This lack of therapeutic lifestyle benefit is consistent with the understanding that Lp(a) levels are genetically determined with plasma concentrations remaining relatively stable over an individual’s lifetime. *Plasma apheresis the most effective way to decrease Lp(a) levels , although, due to cost and technical difficulties, apheresis is generally reserved for severe cases of hyperlipidemia. *A number of pharmacologic agents will lower Lp(a) levels, although compared to other lipid abnormalities, effective drug treatment for elevated Lp(a) levels is relatively limited. *The effect of statins on Lp(a) concentration is variable and also varies with the statin used. It is not clear by what molecular mechanism various statins lower, fail to lower, or even elevate Lp(a) levels. Baseline Lp(a) concentrations and/or apo(a) isoform sizes may influence any statin effect. * Niacin reduces Lp(a) levels by up to 30-40% in dose dependent manner(1-3g/day). While there are no direct experimental data explaining the mechanism for its Lp(a)-lowering effects, at present available data points to niacin as the most effective drug option available to address Lp(a) elevation. *Niaspan, a prescription extended-release (ER) formulation of nicotinic acid has been effectively used in doses of 1-3 gm/day to treat patients with mixed hyperlipidemia and elevated plasma Lp(a) levels. In response to Niaspan treatment there was a substantial benefit with a very favorable improvement in the lipid profile. Lp(a) in type 2 diabetes mellitus several studies have examined the possibility that type 2 diabetes could influence Lp(a) concentrations. The results of several small case–control studies have been controversial. Subjects with diabetes have been found to have higher , similar or lower levels of Lp(a) than controls without diabetes. A recent prospective study of healthy US women aged 45 years or older revealed an inverse association between Lp(a) and the risk of incident type 2 diabetes. The authors replicated their findings in a Danish population-based cohort with prevalent diabetes. These findings suggest that Lp(a) has opposite effects on the risks of cardiovascular disease and diabetes, increasing the former and decreasing the latter. Several population-based studies conducted after adjusting for established risk factors for diabetes (age, abdominal obesity, blood pressure, and serum levels of HDL cholesterol and triglycerides), the age-related increase in the probability of having diabetes was significantly lower in subjects with higher levels of Lp(a). Because both Lp(a) and diabetes increase the risk of cardiovascular disease, mortality rates may be increased at earlier ages in subjects with both risk factors. However, this possibility is inconsistent with recent data showing that, in contrast to what is observed in the general population, plasma Lp(a) levels are not significantly associated with cardiovascular risk in patients with diabetes It has been found that the age-related rate of progression to diabetes is slower in subjects with high levels of Lp(a). Specifically, the age-related risk of diabetes was lower for subjects with Lp(a) levels above 46 mg/dl. The values of HOMA-IR, a surrogate marker of insulin resistance were lower in subjects with Lp(a) levels >46 mg/dl, suggesting that extremely high levels of Lp(a) are associated with less resistance to insulin. Also a positive correlation was found between insulin sensitivity and Lp(a) levels in normoglycemic subjects. Other study demonstrated that Lp(a) levels are inversely correlated with fasting insulin levels and with insulin and glucose concentrations measured 2 hours after an OGTT . Several studies have shown a positive correlation between Lp(a) and HDL cholesterol and a negative correlation between Lp(a) and triglycerides , indicating that Lp(a) shows an inverse relationship with the atherogenic dyslipidemia characteristically associated with insulin resistance. There is no obvious explanation for this inverse correlation between Lp(a) and both diabetes and insulin resistance. Because Lp(a) levels are determined mainly by genetic mechanisms, one possibility is that genetic polymorphisms associated with increased levels of Lp(a) are in linkage disequilibrium with gene(s) that protect against insulin resistance. Thank you