Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Bond valence method wikipedia , lookup
Cluster chemistry wikipedia , lookup
Hydroformylation wikipedia , lookup
Evolution of metal ions in biological systems wikipedia , lookup
Metal carbonyl wikipedia , lookup
Metalloprotein wikipedia , lookup
Stability constants of complexes wikipedia , lookup
Jahn–Teller effect wikipedia , lookup
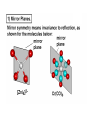
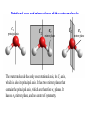
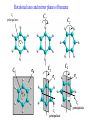
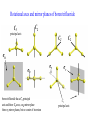
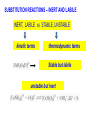
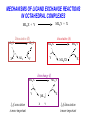
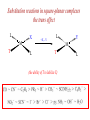
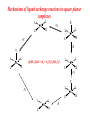
COORDINATION CHEMISTRY STRUCTURES AND ISOMERS ELECTRONIC CONFIGURATION Ground State: Progressive filling of the 3d, 4d, and 5d orbitals Exceptions: ns1 (n-1)d5 rather than ns2 (n-1)d4 ns1 (n-1)d10 rather than ns2 (n-1)d9 Transition metal ions: First in first out TRENDS - IONIC Radii COORDINATION COMPOUNDS Coordination compounds – compounds composed of a metal atom or ion and one or more ligands. [Co(Co(NH3)4(OH2)3]Br6 Ligands usually donate electrons to the metal Includes organometallic compounds Werner’s totally inorganic optically active compound. WERNER’S COORDINATION CHEMISTRY • Performed systematic studies to understand bonding in coordination compounds. – Organic bonding theory and simple ideas of ionic charges were not sufficient. • Two types of bonding – Primary – positive charge of the metal ion is balanced by negative ions in the compound. – Secondary – molecules or ion (ligands) are attached directly to the metal ion. • Coordination sphere or complex ion. • Look at complex on previous slide (primary and secondary) WERNER’S COORDINATION CHEMISTRY • He largely studied compounds with four or six ligands. – Octahedral and square-planar complexes. • It was illustrated that a theory needed to account for bonds between ligands and the metal. – The number of bonds was commonly more than accepted at that time. • 18-electron rule. • New theories arose to describe bonding. – Valence bond, crystal field, and ligand field. COORDINATION COMPOUNDS COORDINATION COMPOUNDS LIGANDS LIGANDS LIGANDS CHELATING LIGANDS Chelating ligands (chelates) – ligands that have two or more points of attachment to the metal atom or ion. Bidentate, tridentate, tetra.., penta…, hexa… (EDTA). trisoxalatochromate(III) ion or just [Cr(ox)3]3- A HEXADENTATE LIGAND, EDTA • There are six points of attachment to the calcium metal. – Octahedral-type geometry ethylene diamine tetraacetic acid (EDTA) ethylenediaminetetraacetatocalcium ion or just [Ca(EDTA)]2- LIGANDS NOMENCLATURE • Coordination compounds that are ionic, the cation is named first and separated by a space from the anion, as is the case for all ionic compounds. The names of neutral coordination complexes are written without spaces. Na[PtCl3(NH3)] Sodium amminetrichloroplatinate(II) K2[CuBr4] Potassium tetrabromocuprate(II) NOMENCLATURE trans-[Co(en)2I(H2O)](NO3)2 trans-aquabis(ethylenediamine)iodocobalt(III) nitrate mer-[Ru(PPh3)3Cl3] mer-trichlorotris(triphenylphosphine)ruthenium(III) NOMENCLATURE • The name of the coordination compound (neutral, cationic or anionic) begins with the names of the ligands. The metal is listed next, following in parentheses by the oxidation state of the metal. NOMENCLATURE When more than one of a given ligand is bound to the same metal atom or ion, the number of such ligands is designated by the following prefixes: 2 di 6 hexa 10 deca 3 tri 7 hepta 11 undeca 4 tetra 8 octa 12 dodeca 5 penta 9 nona NOMENCLATURE However, when the name of the ligand in question already contains one of these prefixes or ligands with complicated names (generally ligand names that are three syllables or longer), then a prefix from the following list is used instead: 2 bis 6 hexakis 3 tris 7 heptakis 4 tetrakis 8 octakis 5 pentakis 9 ennea NOMENCLATURE Neutral ligands are given the same name as the uncoordinated molecule, but with spaces omitted. Some examples are: (CH3)3SO dimethylsulfoxide (DMSO) (NH2)2CO urea C5H5N pyridine terpy terpyridine bpy 2,2’-bipyridine en ethylenediamine PCl3 trichlorophosphine PPh3 triphenylphopshine NOMENCLATURE EXCEPTIONS: Some neutral molecules, when serving as ligands are given special names. These are: NH3 ammine H2O aqua NO nitrosyl CO carbonyl CS thiocarbonyl NOMENCLATURE • Anionic ligands are given names that end in the letter “o”. When the name of the free, uncoordinated anion ends in “ate”, the ligand name is changed to end in “ato”. Some examples are : CH3CO2- (acetate) acetato SO42- (sulfate) sulfato CO32- (carbonate) carbonato acac acetylacetonato NOMENCLATURE When the name of the free, uncoordinated anion ends in “ide”, the ligand name is changed to end in “ido”. Some examples are: N3- (nitride) nitrido N3- (azide) azido NH2- (amide) amido NOMENCLATURE When the name of the free, uncoordinated anion ends in “ite”, the ligand name is changed to end in “ito”. Some examples are: NO2- (nitrite) nitrito SO32- (sulfite) sulfito ClO3- (chlorite) chlorito NOMENCLATURE Certain anionic ligands are given special names, all ending in “o”: CNcyano Ffluoro Clchloro Brbromo Iiodo O2oxo O2superoxo OHhydroxo Hhydrido CH3O- methoxo NOMENCLATURE The ligands are named alphabetically, ignoring the prefixes bis, tris, etc… NOMENCLATURE When the coordination entity is either neutral or cationic, the usual name of the metal is used, followed in parentheses by the oxidation state of the metal. However, when the coordination entity is an anion, the name of the metal is altered to end in “ate”. This is done for some metals by simply changing the ending “ium” to “ate”: Scandium scandate Titanium titanate Chromium chromate NOMENCLATURE Geometrical isomers are designated by cis- or trans- and mer- or fac-, the latter two standing for meridional or facial, respectively. NOMENCLATURE Bridging ligands are designated with the prefix -. When there are two bridging ligands of the same kind, the prefix di-- is used. Bridging ligands are listed in order with other ligands, and set off between hypens. An important exception arises when the molecule is symmetrical, and a more compact name can be given by listing the bridging ligand first. NOMENCLATURE Example: NOMENCLATURE NOMENCLATURE NOMENCLATURE NOMENCLATURE Ligands that are capable of linkage isomerism are given specific names for each mode of attachment. -SCNthiocyanato (S-thiocyanato) -NCSisothiocyanto (N-thiocyanto) -NCSeisoselenocyanato (N-selenocyanato) -NO2nitro -ONOnitrito EXAMPLES 1. 2. 3. 4. 5. 6. 7. [Co(NH3)5CO3]Cl Potassium pentachloronitridoosmate(VI) [Cr(H2O)4Cl2]Cl Potassium pentacyanonitrosylferrate(II) K4[Mn(CN)6] [Ni(bipy)3(NO3)2] [Co(N3)(NH3)5]SO4 NOMENCLATURE • Bridging ligands between two metal ions have the prefix ‘ ’. – -amido--hydroxobis(tetraamminecobalt)(IV) ISOMERISM ISOMERISM ISOMERISM • Four-coordinate complexes – Square-planar complexes may have cis and trans isomers. No chiral isomers (enantiomers) are possible when the molecule has a mirror plane. – cis- and transdiamminedichloroplatinum(II) – How about tetrahedral complexes? – Chelate rings commonly impose a ‘cis’ structure. Why ISOMERISM ISOMERISM CHIRALITY • Mirror images are nonsuperimposable. • A molecule can be chiral if it has no rotation-reflection axes (Sn) • Chiral molecules have no symmetry elements or only have an axes of proper rotation (Cn). – CBrClFI, Tetrahedral molecule (different ligands) – Octahedral molecules with bidentate or higher chelating ligands – Octahedral species with [Ma2b2c2], [Mabc2d2], [Mabcd3], [Mabcde2], or [Mabcdef] CHIRALITY SIX-COORDINATE OCTAHEDRAL COMPLEXES ML3L3’ Fac isomers have three identical ligands on the same face. Mer isomers have three identical ligands in a plane bisecting the molecule. ISOMERISM SIX-COORDINATE OCTAHEDRAL COMPLEXES The maximum number of isomers can be difficult to calculate (repeats). Placing a pair of ligands in the notation <ab> indicates that a and b are trans to each other. [M<ab><cd><ef>], [Pt<pyNH3><NO2Cl><BrI>] How many diastereoisomers in the above platinum compound (not mirror images)? Identify all isomers belonging to Ma3bcd. COMBINATIONS OF CHELATE RINGS • Propellers and helices – Left- and right-handed propellers • Examine the movement of a propeller required to move it in a certain direction. – For a left-handed propeller, rotating it ccw would cause it to move away (). – For a right-handed propeller, rotating it cw would cause it to move away (). This is called ‘handedness’. Many molecules possess it. Tris(ethylenediamine)cobalt(III) • this molecule can be treated like a three-bladed propeller. • look down a three fold axis to determine the ‘handedness’ of this complex ion. – the direction of rotation required to pull the molecule away from you determines the handedness ( or ). • do this with you molecule set and rubber bands. DETERMINING HANDEDNESS FOR CHIRAL MOLECULES • Complexes with two or more nonadjacent chelate rings may have chiral character. – Any two noncoplanar and nonadjacent chelate rings can be used. – Look at Figure 9-14 (Miessler and Tarr). • Molecules with more than one pair of rings may require more than one label. – Ca(EDTA)2+ • Three labels would be required. • Remember that the chelate rings must be noncoplanar, nonadjacent, and not connected at the same atom. LINKAGE (AMBIDENTATE) ISOMERISM A few ligands may bond to the metal through different atoms. SCN- and NO2 How would you expect hard acids to bond to the thiocyanate ligand? Solvents can also influence bonding. High and low dielectric constants. Steric effects of linkage isomerism Intramolecular conversion between linkages. [Co(NH3)5NO2]+2, Figure 9-19. COORDINATION NUMBERS AND STRUCTURES • Factors considered when determining structures. – The number of bonds. Bond formation is exothermic; the more the better. – VSEPR arguments – Occupancy of d orbitals. – Steric interference by large ligands. – Crystal packing effect. It may be difficult to predict shapes. LOW COORDINATION NUMBERS (C.N.) • CN 1 is rare except in ion pairs in the gas phase. • CN 2 is also rare. – [Ag(NH3)2]+, Ag is d10 (how?) – VSEPR predicts a linear structure. – Large ligands help force a linear or near-linear arrangment. • [Mn(N[SiMePh2]2)2] in Figure 9-22. • C.N. 3 is more likely with d10 ions. – Trigonal-planar structure is the most common. – [Cu(SPPh3)3]+, adopts a low C.N. due to ligand crowding. COORDINATION NUMBER 4 • Tetrahedral and square planar complexes are the most common. – Small ions and/or large ligands prevent high coordination numbers (Mn(VII) or Cr(VI)). • Many d0 or d10 complexes have tetrahedral structures (only consider bonds). – MnO4- and [Ni(CO)4] – Jahn-Teller distortion (Chapter 10) COORDINATION NUMBER 4 • Square-planar geometry – d8 ions (Ni(II), Pd(II), and Pt(III)) • [Pt(NH3)2Cl2] – The energy difference between square-planar and tetrahedral structures can be quite small. • Can depend on both the ligand and counterion. • More in chapter 10. COORDINATION NUMBER 5 • Common structures are trigonal bipyramid and square pyramid. – The energy difference between the two is small. In many measurements, the five ligands appear identical due to fluxional behavior. – How would you modify the experiment to differentiate between the two structures? • Five-coordinate compounds are known for the full range of transition metals. – Figure 9-27. COORDINATION NUMBER 6 • This is the most common C.N. with the most common structure being octahedral. – If the d electrons are ignored, this is the predicted shape. • [Co(en)3]3+ • This C.N. exists for all transition metals (d0 to d10). DISTORTIONS OF COMPLEXES CONTAINING C.N. 6 • Elongation and compression (Fig. 9-29). – Produces a trigonal antiprism structure when the angle between the top and bottom triangular faces is 60. – Trigonal prism structures are produced when the faces are eclipsed. • Most trigonal prismatic complexes have three bidentate ligands (Figure 9-30). • interactions may stabilize some of these structures. The Jahn-Teller effect is useful in predicting observed distortions. HIGHER COORDINATION NUMBERS • C.N. 7 is not common • C.N. 8 – There are many 8-coordinate complexes for large transition elements. • Square antiprism and dodecahedron • C.N.’s up to 16 have been observed. MAGNETIC SUSCEPTIBILITY • Diamagnetic versus paramagnetic complexes. • Commonly provides mass susceptibility per gram. • Magnetic moment 2.828( T ) 1 2 magnetic susceptibility CONTRIBUTIONS TO THE MAGNETIC MOMENT • Spin magnetic moment – S = maximum total spin in the complex • O atom • Orbital angular momentum – Characterized by the quantum number L which is equal the maximum possible sum of ml values. • O atom S L g [ S ( S 1)] [ 1 4 L( L 1)] CONTRIBUTIONS TO THE MAGNETIC MOMENT • Usually, the spin-only moment is sufficient to calculate the magnetic moment. – Especially for the first transition series S g S(S 1) or n(n 2) where g (gyromagnetic ratio) is approximated to be 2 and n is the number of unpaired electrons. – Determine the spin-only and complete magnetic moment for Fe. Calculate the spin-only magnetic moment For the following atoms/ions: Fe+2 (observed: 5.1), Fe, Cr, Cr+3 (observed = 3.8) ELECTRONIC SPECTRA • Orbital energy levels can be obtained directly from electron spectra (will be covered later). • This chapter illustrates simple energy level diagrams that are commonly more complex. • Based upon subtle differences in electronic spectra, the structure may be predicted with some success. THEORIES OF ELECTRONIC STRUCTURE • Valence Bond Theory – Not commonly used, but the hybrid notation is still common. • Crystal Field Theory – An electrostatic approach used to describe the splitting in metal d-orbital energies. Does not describe bonding. • Ligand Field Theory – A more complete description of bonding in terms of the electronic energy levels of the frontier orbitals. Commonly does not include energy of the bonding orbitals. • Angular Overlap Method – Used to estimate the relative magnitude of the orbital energies in a MO calculation. VALENCE BOND THEORY (HYBRIDIZATION) • A set of hybrid orbitals is produced to explain the bonding. – Octahedral – d2sp3 (6 hybrid orbitals of equal energy) – Tetrahedral - ?? • Uses ‘inner’ and ‘outer’ orbitals to explain the experimentally determined unpaired electrons. – The magnetic behavior determines which d orbitals (e.g. 3d or 4d) are used for bonding (Figure 10-2). VALENCE BOND DESCRIPTION • Two configurations are possible for d4-d7 ions. • Fe(III) has 5 electrons in the d-orbitals. – One unpaired electron, the ligands are ‘strong’ and force the metal d electrons to pair up. • Strong-field (bind strongly) low spin complex • The hybridization orginates from the 3d inner orbitals (d2sp3). VALENCE BOND DESCRIPTION – Five unpaired electrons, the ligands are ‘weak’ and cannot force the metal d electrons to pair up. • Weak-field (bind weakly) high spin • The hybridization originates from the 4d outer orbitals (sp3d2). VALENCE BOND THEORY Structure, hybridization, and magnetism 1) [Co(NH3)6]3+, diamagnetic, octahedral 2) [CoF6]3-, paramagnetic, octahedral 3) [PtCl4]2-, diamagnetic, sq. planar 4) [NiCl4]2-, pamagnetic, tetrahedral SAMPLE PROBLEM: • The complexes [Mn(H2O)6]2+, [Fe(H2O)6]3+, [MnCl4]2-, and [FeCl4]- have all magnetic moments. What does this tell about the geometric and electronic structures of these complexes? CRYSTAL FIELD THEORY • Focus: energies of the d orbitals • Assumptions 1. Ligands: negative point charges 2. Metal-ligand bonding: entirely ionic strong-field (low-spin): large splitting of d orbitals weak-field (high-spin): small splitting of d orbitals 20_454 eg(d z2, d x 2 – y 2) t2g (d xz, d yz, d xy) E = crystal field splitting Free metal ion 3d orbital energies High spin Low spin CRYSTAL FIELD THEORY • The average energy of the d-orbitals in the presence of the octahedral field is greater than than of the free ion. • Energy difference between the two sets is equal to O. – The t2g set is lowered by 0.4 O and the eg set is raised by 0.6 O. • Crystal field stabilization energy (CFSE) – The energy difference between the actual distribution of electrons and that for all electrons in the uniform field. – Equal to LFSE (later) • Drawbacks LIGAND FIELD THEORY – OCTAHEDRAL COMPLEXES • Consider -type bonding between the ligands and the metal atom/ion. • Construct LGOs (performed previously). – What is the reducible representation? – Construct the LGOs (pictures). • Construct the molecular orbitals with the metal orbitals. – Same symmetry types. • A group of metal orbitals do not have the appropriate symmetry? – Which orbitals are these? Symmetry type? Bonding? • Look at Figure 10-5. SF6 = A1g + T1u + Eg LIGAND FIELD THEORY – OCTAHEDRAL COMPLEXES • The six bonding orbitals are largely filled by the electrons from the ligands. • The higher MOs (e.g. t2g and eg) are largely filled by the electrons on the metal atom/ion. – The ligand field treatment largely focuses on the t2g and higher orbitals. • The split between the two sets of orbitals, t2g and eg, is called O. LIGAND FIELD THEORY – OCTAHEDRAL COMPLEXES • Ligands whose orbitals interact strongly with the metal orbitals are called strong-field ligands. – Strong-field large O low spin (why?) • Ligands with small interactions are called weak-field ligands. – Weak-field small O high spin (why?) • For d0 – d3 and d8-d10 only one electron configuration is possible (no difference in net spin). • For d4 – d7 there is a difference between strong- and weakfield cases. LOW SPIN VERSUS HIGH SPIN • Energy of pairing electrons c e – c is the Coulombic energy of repulsion (always positive when pairing) and e is the quantum mechanical exchange energy (always negative). • e relates to the number of exchangeable pairs in a particular electron configuration. This term is negative and depends on the number of possible states. Determine c and e for a d5 metal complex (low and high spin). LOW SPIN VERSUS HIGH SPIN • The relationship between O, c, and e determines the orbital configuration. • is largely independent on the ligands while O is strongly dependent. • Look at Table 10-6 which gives these parameters for aqueous (aqua) ions. – O for 3+ ions is larger than O for 2+ ions. – O values for d5 are smaller than d4 and d6. LOW SPIN VERSUS HIGH SPIN • If O>, there is a lower energy upon pairing in the lower levels (low spin). • If O<, there is a lower energy with unpaired electrons in the lower levels (high spin). • In Table 10-6, [Co(H2O)6]3+ is probably the only complex that could be low spin. Ligand Field Stabilization Energies (LFSE) • The difference (1) the total energy of a coordination complex with the electron configuration resulting from ligand field splitting of the orbitals and (2) the total energy for the same complex with all the orbitals equally populated is the LFSE. • -2/5O + 3/5O (d4 to d7 complexes) • Table 10-7 BONDING IN OCTAHEDRAL COMPLEXES • The x and z axes must be taken as a single set producing a combined LGO set. Why? • Be able to derive the reducible representation. – = T1g + T2g + T1u + T2u • How will the LGOs combine with orbitals from the metal atom/ion? • Discuss the overlap between the -bonding LGOs and the p-orbitals of T1u symmetry. PI BONDING IN OCTAHEDRAL COMPLEXES • The main addition to the interaction diagram is between the t2g orbitals of the metal and LGOs. – These were nonbonding when only considering type bonding (look at Figure 10-5). • Pi bonding may occur when the ligands have available p or * molecular orbitals. LIGANDS WITH EMPTY * ORBITALS • Examine the example for the CN- ligand in the book (Figure 10-9). • The HOMO forms the LGOs from -type bonding (already discussed previously). • The LUMO, 1*, also forms a reducible set of LGOs (T1g + T2g + T1u + T2u). – Examine Figure 10-10 to illustrate effectiveness of overlap. LIGANDS WITH EMPTY * ORBITALS • The resulting t2g LGOs are generally higher in energy than the initial t2g orbitals on he metal. – Bonding/antibonding t2g orbitals will result. – What will this do to O and the bond strength? • Figure 10-11. • This is termed as metal-to-ligand bonding or back-bonding. – Some of the electron density in the d orbitals on the metal is donated back to the ligands. – The ligands are termed as -acceptor ligands. LIGANDS WITH FILLED -TYPE ORBITALS • Ligands such as F- or Cl- will possess molecular orbitals that possess electrons. • This set of ‘t2g’ orbitals are generally lower in energy than the t2g orbitals on the metal. • What are the consequences? – Examine Figure 10-11. • Ligand-to-metal bonding (-donor ligands). – This bonding is generally less favorable. SQUARE-PLANAR COMPLEXES • The y-axis is pointed toward the center atom. – LGOs for sigma-type bonding. • The -bonding orbitals on the x- and z-axes have to be considered separately? Why? – These are termed as (px) and (pz) • Examine Table 10-9. – What is the symmetry of a square-planar complex? SQUARE-PLANAR COMPLEXES SIGMA-TYPE BONDING ONLY • Finding the LGOs. – red = A1g + B1g + Eu • What are the orbitals on the central metal atom that can interact with these LGOs? • Inspecting the character table reveals that the metal d-orbitals are split into three representations. Why? • Examine Figure 10-13. – The energy difference between the eg/b2g nonbonding orbitals and the a1g antibonding is . SQUARE-PLANAR COMPLEXES INCLUDING PI-BONDING • px = A2g + B2g + Eu () – What are the interacting orbitals on the metal? • pz = A2u + B2u + Eg () – What are the interacting orbitals on the metal? • The effective overlap of the p orbitals on the metal to form bonds is small. • Examine Figure 10-15. THE ‘SETS’ OF ORBITALS IN FIGURE 10-15 • The 1st set contains bonding orbitals (mostly sigma). – 8 electrons from the ligands largely fill these orbitals. • The 2nd set contains 8 -donor orbitals of the ligands. – This interaction is small and decreases the energy differences in orbitals the next higher set. • The 3rd set is primarily metal d-orbitals with some modifications due to interactions with the ligands. – 3, 2, and 1 are in this set. • The 4th set largely originates from the * orbitals of the ligands (if present). – One of the main effects of these orbitals is the increase in the gap energy labeled 1. ANGULAR OVERLAP (CRYSTAL FIELD) • Estimates the strength of interaction between individual ligand orbitals and d-orbitals based on the overlap between them. These values are then combined for all ligands and d-orbitals. • The value for a given d-orbital is the sum of the numbers for the appropriate ligands in a column. – This number can be positive or negative depending on location of the ligand and d-orbitals. • The value for a given ligand is the sum of the numbers for all dorbitals in the row. – This number can also be positive or negative depending on location of the ligand and d-orbitals. ANGULAR OVERLAP • Sigma-donor interaction (no pi-orbitals are available). – [M(NH3)6]n+ • The strongest interaction is between the metal dz2 orbital and a ligand p-orbital (or appropriate MO). • Describe the interaction based on this method. ANGULAR OVERLAP • Pi-acceptor ligands (available -type orbitals). • Strongest interaction is between dxz and * on the ligand. • The * orbitals are almost always higher in energy. – Reverse the signs. • Figure 10-22 and Table 10-12 – There is a lowering of 4e due to this interaction. • Why is magnitude e always smaller than that of e? • Understand -donor interactions. SAMPLE PROBLEM Using the angular overlap model, determine the splitting pattern of the d orbitals for a tetrahedral complex of formula ML4.where L is a capable of interactions only. SAMPLE PROBLEM Determine the energies of the d orbitals predicted by the angular overlap model for square planar complexes a) considering interactions only b) considering both -donor and acceptor interactions THE SPECTROCHEMICAL SERIES • depends on the relative energies and the degree of overlap. • How ligands effect – -donor ligands – -donating – -accepting (or back bonding) • Understand the spectrochemical series (page 368) MAGNITUDE OF E, E, AND • Changing the metal and/or ligand effects the magnitudes of e and e, thereby changing the value of . – Aqua species of Co2+ and Co3+ – [Fe(H2O)6]2+ versus [Fe(H2O)6]3+ • Tables 10-13 and 10-14 (Angular Overlap) – e > e (always) – Values decrease with increasing size and decreasing electronegativity – Negative values for e. Why? THE JAHN TELLER EFFECT • There cannot be unequal occupation of orbitals with identical energies. The molecule will distort so that these orbitals are no longer degenerate. – Cu(II) d9 ion, The complex will distort. How? – The low-spin Cr(II) complex is octahedral with tetragonal distortion (Oh D4h) • Two absorption bands are observed instead of one. DETERMINING FOUR- AND SIX-COORDINATE PREFERENCES • General angular overlap calculations of the energies expected for different number of d electrons and different geometries can give us some indication of relative stabilities. – Larger number of bonds usually make the octahedral complexes more stable. Why are the energies equal in the d5, d6, and d7 cases? – Figure 10-27. DETERMINING FOUR- AND SIX-COORDINATE PREFERENCES • The success of these simplistic calculations is variable. – The s- and p-orbitals of the metal are not included. – No -type interactions are included in Figure 10-27. – The orbital potential energies for the metals change with increasing atomic number (more negative). • Can add –0.3e (increase in Z) as a rough correction to THE PROCESS FOR A COMPLEX OF D3h SYMMETRY • Construct the sigma-type bonding LGOs for the complex. • Determine the interacting orbitals on the center atom. • Construct a table to determine e (and e if appropriate). • Construct the MO diagram and overlap energy figure. Homework: Determine the e contribution. Symmetry and Group Theory The symmetry properties of molecules and how they can be used to predict vibrational spectra, hybridization, optical activity, etc. POINT GROUPS Molecules are classified and grouped based on their symmetry. Molecules with similar symmetry are but into the same point group. A point group contains all objects that have the same symmetry elements. SYMMETRY ELEMENTS Symmetry elements are mirror planes, axis of rotation, centers of inversion, etc. A molecule has a given symmetry element if the operation leaves the molecule appearing as if nothing has changed (even though atoms and bonds may have been moved.) Element n-fold axis Mirror plane σ Center of inversion n-fold axis of improper rotation Symmetry Operation Symmetry Elements Identity Rotation by 2π/n Reflection Inversion Rotation by 2π/n followed by reflection perpendicular to the axis of rotation Symbol E Cn i Sn C3 C3 or three-fold rotational axis of the ammonia molecule. If we rotate the ammonia molecule by 360/3 or 120º about this axis, its appearance is unchanged. Rotational axes of BF3 principal axis (highest value of Cn) C3 C3 C2 C2 . three-fold axis viewed from above three-fold axis viewed from the side two-fold axis viewed from the side two-fold axis viewed from above Note: there are 3 C2 axes SAMPLE PROBLEM • How many axes of rotation does borazine possess? • Ethane in the eclipsed conformation? Mirror planes (σ) of BF3: Mirror planes can contain the principal axis (σv) or be at right angles to it (σh). BF3 has one σh and three σv planes: (v = vertical, h = horizontal) σv mirror plane σv mirror plane contains the C3 axis C3 principal axis σh mirror plane C3 principal axis σh mirror plane is at right angles to the C3 axis SAMPLE PROBLEM • Mirror planes of symmetry for Borazine, naphtlalene, diborane, dxy orbital? center of symmetry center of symmetry (Note: The center of symmetry is important in deciding whether orbitals are g or u (lecture 2.)) SAMPLE PROBLEM • Which of the following flourine compounds has center of inversion? BF3, SiF4, PF5, XeF5-, SF6, C2F4, rotate by 360o/4 The S improper rotation axis here is also a C axis Rotational axes and mirror planes of the water molecule: C2 principal axis C2 σv mirror plane The water molecule has only one rotational axis, its C2 axis, which is also its principal axis. It has two mirror planes that contain the principal axis, which are therefore σv planes. It has no σh mirror plane, and no center of symmetry. C2 σv mirror plane Rotational axes and mirror planes of benzene C6 C2 principal axis C2 C6 σh C2 C2 σv C6 principal axis σv C6 principal axis Rotational axes and mirror planes of boron trifluoride C2 C3 principal axis C2 C2 σh σh boron trifluoride has a C3 principal axis and three C2 axes, a σh mirror plane three σv mirror planes, but no center of inversion σv σv C3 principal axis Identity, E All molecules have Identity. This operation leaves the entire molecule unchanged. A highly asymmetric molecule such as a tetrahedral carbon with 4 different groups attached has only identity, and no other symmetry elements. Improper Rotation An improper rotation is rotation, followed by reflection in the plane perpendicular to the axis of rotation. Improper Rotation The staggered conformation of ethane has an S6 axis that goes through both carbon atoms. Improper Rotation Note that an S1 axis doesn’t exist; it is same as a mirror plane. Improper Rotation Likewise, an S2 axis is a center of inversion. Sample problem Draw the structure for the following showing the correct geometry and identify all the symmetry elements present in each: a) SCN- b) S2O32-, c) IF4- d) 1,8-dichloronaphthalene e) formaldehyde Point Groups Molecules with the same symmetry elements are placed into point groups. Group theory, the mathematical treatment of the properties of groups can be used to determine the molecular orbitals, vibrations, and other properties of the molecule. ∞ ∞ Point Groups In general, you will not need to assign a molecule to its point group. Recognition of the features of some common point groups is useful. Point Groups Water and ammonia both belong to the Cnv class of molecules. These have vertical planes of reflection, but no horizontal planes. Point Groups The Dnh groups have a horizontal plane in addition to vertical planes. Many inorganic complexes belong to these symmetry groups. Y X X X X Y POINT GROUPS Highly symmetrical molecules, such as identically substituted tetrahedrons or octahedrons belong to their own point groups (Td or Oh respectively). Point Groups In assigning a point group, we typically ignore the fine detail, such as conformation isomers, of the ligands. In working problems using group theory, the point group of the molecule will usually be provided to you. Example: • PF5, SF6, IOF3, XeF4, ethane (eclipsed and staggered), ethylene and chloroethylene. Ferrocene (eclipsed and staggered) COORDINATION CHEMISTRY III: REACTIONS OF METAL COMPLEXES The ability to predict products and choose appropriate reaction condition to obtain the desired products is still a matter of art as well as science. substitution reactions kinetic consequences of reaction pathways experimental evidence in octahedral substitution substitution reactions of square-planar complexes the trans effect oxidation-reduction reactions reactions of coordinated ligand SUBSTITUTION REACTIONS SUBSTITUTION REACTIONS MLn-1L' + L MLn + L' Labile complexes <==> Fast substitution reactions (< few min) Inert complexes <==> Slow substitution reactions (>h) a kinetic concept Not to be confused with stable and unstable (a thermodynamic concept Gf <0) Inert Intermediate d3, low spin d4-d6& d8 d8 (high spin) Labile d1, d2, low spin d4-d6& d7-d10 SUBSTITUTION REACTIONS – INERT AND LABILE INERT, LABILE vs STABLE, UNSTABLE kinetic terms thermodynamic terms Stable but labile unstable but inert MECHANISMS OF LIGAND EXCHANGE REACTIONS IN OCTAHEDRAL COMPLEXES MLnY + X MLnX + Y Dissociative (D) MLn X X Associative (A) MLn Y MLn Y Y MLn X Interchange (I) Y Ia if association is more important MLn Y MLn X [ML n]° X Y MLn XY MLn Y X Id if dissociation is more important X KINETICS OF DISSOCIATIVE REACTIONS Kinetics of interchange reactions Fast equilibrium K1 = k1/k-1 k2 << k-1 For [Y] >> [ML5X] Kinetics of associative reactions Principal mechanisms of ligand exchange in octahedral complexes Dissociative Associative Dissociative pathway (5-coordinated intermediate) MOST COMMON Associative pathway (7-coordinated intermediate) Experimental evidence for dissociative mechanisms Rate is independent of the nature of L Experimental evidence for dissociative mechanisms Rate is dependent on the nature of L Inert and labile complexes Some common thermodynamic and kinetic profiles Exothermic (favored, large K) Large Ea, slow reaction Exothermic (favored, large K) Large Ea, slow reaction Stable intermediate Endothermic (disfavored, small K) Small Ea, fast reaction Labile or inert? L L L M L L Ea L L L L M L L M L L L X L X G LFAE = LFSE(sq pyr) - LFSE(oct) Why are some configurations inert and some are labile? Inert ! Other metal on factors that affect reaction rates Oxidation state of the central atom: Central atom with higher oxidation states have slower ligand exchange rates [AlF6]- > [SiF6]- > [PF6]- > SF6 Ionic radius. Smaller ions have slower exchange rates [Sr(H2O)6]2+ > [Ca(H2O)6]2+ > [Mg(H2O)6]2+ 112 pm 99 pm 66 pm Both effects due to higher electrostatic attraction between central atom and attached ligands. Substitution reactions in square-planar complexes the trans effect L X M T L +X, -Y L Y M T (the ability of T to labilize X) L Synthetic applications of the trans effect Mechanisms of ligand exchange reactions in square planar complexes L L X L S +S M L L M X L +Y -X Y L L L -d[ML3X]/dt = (ks + ky [Y]) [ML3X] M X L L M S L +Y Y L -X L L L L M Y -S L M S THE trans EFFECT SIGMA-BONDING EFFECTS Sigma-Bonding Effect. A strong bond between Pt and T weakens the Pt-X bond. H- > PR3 > SCN- ~ CH3- ~ CO ~ CN- > Br- > Cl- > NH3 > OH- PI-BONDING EFFECTS If back donation occurs to a ligand, the flow of electron density from the metal leaves less electron density to be donated in the opposite direction. C2H4 ~ CO > CN- > NO2- > SCN- > I- > Br- > Cl- > NH3 > OH- Overall trans effect: CO ~ CN- ~ C2H4 > PR3 ~ H- > CH3- ~ SC(NH2)2 > C6H5>NO2- ~ SCN ~ I- >Br- > Cl- > py , NH3 ~ OH- ~H2O SAMPLE PROBLEM: Predict the products of the reactions (there may be one product when there are conflicting preferences) [PtCl4-] + NO2- → (a) [PtCl3NH3]- + O2- → (c) (a) + NH3 → (b) (c) + NO2- → (d) SAMPLE PROBLEM: Is it possible to prepare different isomers of Pt(II) complexes with 4 different ligands? Predict the products expected if 1 mole of [PtCl4]- is reacted successively with the following reagents: (the product of reaction a is used in reaction b) a) b) c) d) 2 moles NH3 2 moles py 2 moles Cl1 mole NO2- Electron transfer (redox) reactions -1e (oxidation) M1(x+)Ln + M2(y+)L’n M1(x +1)+Ln + M2(y-1)+L’n +1e (reduction) Very fast reactions (much faster than ligand exchange) May involve ligand exchange or not Very important in biological processes (metalloenzymes) REDOX MECHANISMS: Inner sphere mechanism: When two molecules are connected by a common ligand which the electron is transferred, in which case the reaction is called bridging or innersphere reaction. Outer sphere mechanism: Exchange may occur between two separate coordination sphere in outersphere reaction. Outer sphere mechanism [Fe(CN)6]3- + [IrCl6]3- [Fe(CN)6]4- + [IrCl6]2- [Co(NH3)5Cl]+ + [Ru(NH3)6]3+ [Co(NH3)5Cl]2+ + [Ru(NH3)6]2+ Reactions ca. 100 times faster than ligand exchange (coordination spheres remain the same) A B "solvent cage" r = k [A][B] Ea Tunneling mechanism A + B A' G + B' Inner sphere mechanism [Co(NH3)5Cl)]2+ + [Çr(H2O)6]2+ [Co(NH3)5Cl)]2+:::[Çr(H2O)6]2+ [CoIII(NH3)5(-Cl)ÇrII(H2O)6]4+ [CoII(NH3)5(-Cl)ÇrIII(H2O)6]4+ [CoII(NH3)5(H2O)]2+ [Co(NH3)5Cl)]2+:::[Çr(H2O)6]2+ [CoIII(NH3)5(-Cl)ÇrII(H2O)6]4+ [CoII(NH3)5(-Cl)ÇrIII(H2O)6]4+ [CoII(NH3)5(H2O)]2+ + [ÇrIII(H2O)5Cl]2+ [Ço(H2O)6]2+ + 5NH4+ Inner sphere mechanism Ox-X + Red k1 Ox-X-Red k2 Reactions much faster than outer sphere electron transfer (bridging ligand often exchanged) k3 k4 Ox(H2O)- + Red-X+ Ox-X-Red Tunneling through bridge mechanism r = k’ [Ox-X][Red] k’ = (k1k3/k2 + k3) Ea Ox-X + Red Ox(H 2O) - + Red-X + G