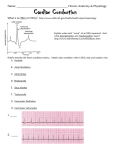
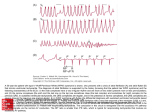
Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
TETC32 12/2/05 14:26 Page 949 32 Ventricular Tachycardia Lars Eckardt, Pedro Brugada, John Morgan and Günter Breithardt Summary Ventricular arrhythmias are the major cause of morbidity and mortality in patients with structural heart disease, but can also be a mechanism of sudden death in patients with structurally normal hearts (e.g. channelopathies such as long or short QT syndrome, Brugada syndrome). Infrequently, they can be generated by mechanisms that are amenable to curative catheter ablation. Overall, ventricular tachycardia and ventricular fibrillation are the major cause of sudden unexpected death. Ventricular tachycardias are relatively organized tachyarrhythmias with discrete QRS complexes. They can be either sustained or nonsustained, and can be monomorphic or polymorphic. Polymorphic ventricular tachyarrhythmias tend to be faster and less stable than monomorphic. The correct diagnosis of a ventricular tachycardia remains a challenge despite numerous established criteria for the Introduction Ventricular arrhythmias are the major cause of morbidity and mortality in patients with structural heart disease, but can also be a mechanism of sudden death in patients with structurally normal hearts. Infrequently they can be generated by mechanisms that are amenable to curative catheter ablation. Overall, ventricular tachycardia (VT) and ventricular fibrillation (VF) are the major cause of sudden unexpected death. Ambulatory ECG recordings at the time of sudden death have shown that, in approximately 60% of sudden cardiac death victims, an episode of VT was identified as the initial event [1].. Major studies of heart failure therapy have shown that ventricular differentiation of ventricular from supraventricular tachycardia with aberrant conduction. A history of heart disease has a positive predictive accuracy of 95% for a ventricular tachyarrhythmia. A re-entry mechanism accounts for the majority of ventricular tachyarrhythmias in patients with structural heart disease. The spectrum of therapies for ventricular tachycardias includes drug therapy, device implantation and surgical or catheter ablation techniques. In patients with chronic coronary heart disease, the magnitude of the survival benefit from the implantable cardioverterdefibrillator is directly related to the severity of cardiac dysfunction. The management challenge is to deal both with the ventricular tachycardia as the presenting symptom, and the pervading sudden cardiac death risk that may be the consequence of the arrhythmogenic substrate. arrhythmia is the commonest cause of death, whatever the functional class of the patient. Definitions Ventricular tachycardia is a relatively organized tachyarrhythmia with discrete QRS complexes. It can be either sustained (lasting longer than 30 s) or non-sustained (defined as three or more beats but less than 30 s), and can be monomorphic or polymorphic. If the same patient has monomorphic ventricular tachycardias with different morphologies, it is termed pleomorphic. Polymorphic 949 TETC32 12/2/05 14:26 Page 950 950 Chapter 32 ventricular tachycardias tend to be faster and less stable than monomorphic. The rate of ventricular tachycardia can range from 100 beats per minute (b.p.m.) to more than 300 b.p.m. At faster rates (usually 220 b.p.m. or faster), ventricular tachycardia is so rapid that it may be impossible to distinguish the QRS complex from the T wave. This type of ventricular tachycardia is referred to as ventricular flutter. Ventricular fibrillation is a completely disorganized (chaotic) tachyarrhythmia without discrete QRS complexes. When it begins, it is associated with a coarse electrical pattern. As the heart becomes less viable, the fibrillation becomes fine, and then, as an agonal event, all electrical activity ceases (flat line). Electrocardiographic diagnosis of ventricular tachycardia The correct diagnosis of a wide complex tachycardia (QRS duration > 120 ms) remains a challenge despite numerous established criteria for the differentiation of ventricular from supraventricular tachycardia with aberrant conduction. Ventricular tachycardia is the most common cause of wide complex tachycardia, accounting for up to 80% of all cases [2]. A history of heart disease (prior myocardial infarction or heart failure) has a positive predictive accuracy of 95% for ventricular tachycardia [3]. On the other hand, if a patient has had similar episodes during previous years, a supraventricular origin is more likely than a ventricular tachycardia. Termination of a tachycardia by physical manoeuvres, such as the Valsalva manoeuvre or adenosine injection, strongly suggests a supraventricular origin, although some ventricular tachycardias can also terminate by these manoeuvres (e.g. fascicular ventricular tachycardia). A wide complex tachycardia in a patient who is alert and haemodynamically stable is not necessarily of supraventricular origin. If it is a ventricular tachycardia in a patient with reduced systolic function, an i.v. injection of, for example, verapamil may result in severe hypotension and haemodynamic instability. In general, if an electrocardiogram (ECG) showing a wide complex tachycardia does not look like aberration, it is most likely a ventricular tachycardia. If there is any doubt about the origin of a broad complex tachycardia, the patient should be treated as if the rhythm is ventricular tachycardia because it is by far the more common diagnosis. The absence of a RS complex in any precordial lead or an interval of the R-wave onset to the S-wave nadir of more than 100 ms strongly suggests a ventricular tachycardia [4]. In addition, the following ECG criteria have been suggested to distinguish between a ventricular and a supraventricular tachycardia with aberration. l QRS complex duration. Ventricular tachycardia is the probable diagnosis when the QRS duration with right bundle branch block (RBBB) is greater than 140 ms, and greater than 160 ms with left bundle branch block (LBBB) morphology [2]. l QRS axis. A frontal axis of between –90 and ±180 degrees cannot be achieved by any combination of bundle branch block and therefore suggests ventricular tachycardia. Thus, predominantly negative QRS complexes in leads I, II and III are useful criteria for identifying a ventricular tachycardia. l Concordant negative ECG patterns in the precordial leads. If all precordial leads are predominantly negative, a ventricular tachycardia is the likely diagnosis. If all precordial leads are predominantly positive, the differential diagnosis is an antidromic tachycardia using a left-sided accessory pathway or a ventricular tachycardia. l QRS morphologies in V1 and V6 (Fig. 32.1). In RBBB pattern, a monophasic R wave, a broad (> 30 ms) R or a QR in V1 strongly suggests ventricular tachycardia. A monophasic R wave or an S greater than an R in V6 LBBB SVT VT small R Slow descent Broad R V1 Fast descent > 60 ms V6 Q RBBB SVT rSR-pattern VT Monophasic R qR (or RS) V1 R/S >1 R/S < 1 or QS pattern V6 Figure 32.1 QRS criteria for differential diagnosis in broad complex tachycardia: ventricular tachycardia (VT) vs. supraventricular tachycardia (SVT) with left (LBBB) or right (RBBB) bundle branch block. TETC32 12/2/05 14:26 Page 951 Ventricular Tachycardia Lead V1 LBBB pattern V1 VT from the RV or LV septum VT from the LV Ischaemic cardiomyopathy = likely LV with septal origin QRS positive V2–V6 = base of LV QRS negative V2–V6 = apex of LV Positive II, III, aVF, normal heart = RVOT-VT II, III, aVF positive = anterior wall II, III, aVF negative = inferior wall QRS > 0.140 s = RVOT free wall QRS < 0.140 s = RVOT septum QS in aVR > aVL = RVOT posterolateral QS in aVR < aVL = RVOT anterior R wave transition before V2 = consider LVOT Figure 32.2 Algorithm for localization of the exit site of ventricular tachycardia. RBBB pattern V1 I and aVL positive = septal wall I and aVL negative = lateral wall Multiple morphologies or negative in II, II, aVF = consider RV-dysplasia also suggests ventricular tachycardia. In the presence of a LBBB pattern, a broad R wave (usually greater than 30 ms [5]) and/or a slow descent to the S wave nadir in V1 and a Q in V6 point towards a ventricular tachycardia. l Atrioventricular dissociation. This is one of the most useful criteria for distinguishing ventricular tachycardia from supraventricular tachycardia (SVT). It occurs in 20–50% of ventricular tachycardia and almost never in SVT [2,6,7]. Atrioventricular dissociation may be diagnosed by a changeable pulse pressure, irregular canon A waves in the jugular veins and a variable first heart sound. It is often very difficult to ascertain, particularly in rapid tachycardias. It often demands long 12-lead ECG recordings and careful ECG analysis. In addition, about 30% of ventricular tachycardias have 1:1 retrograde conduction. In the presence of AV dissociation, one may also observe fusion beat, which may result from the fusion of a P wave conducted to the ventricles. The 12-lead ECG during ventricular tachycardia can be helpful in providing an approximation of the site of origin, which may be helpful for guiding ablation (Fig. 32.2). In general, ventricular tachycardias that have a left bundle branch block-like morphology in V1 have an exit in the right ventricle or the interventricular septum. A QRS axis that is directed superiorly generally indicates an exit in the inferior wall; an axis directed inferiorly indicates an exit in the anterior (superior) wall. In V2–V4, dominant R waves usually indicate an exit near the base of the ventricle. In idiopathic right ventricular outflow tract tachycardia (see RVOT ventricular tachycardia, below), the QRS duration during ventricular tachycardia is usually greater than 140 ms if it originates from the free-wall of the RVOT, and less than 140 ms if the arrhythmia originates from the septal site of the RVOT. Furthermore, if the QS amplitude in aVR is greater than in aVL, the initial activation occurs more posterolateral, whereas if the QS amplitude in aVL is greater than in aVR, the origin is more anterior in the RVOT. The precordial R-wave transition in RVOT-ventricular tachycardia usually occurs in leads V2–V4 and becomes earlier as the site of origin advances more superiorly along the septum. An R-wave transition in lead V2 suggests a site of origin immediately inferior to the pulmonic valve or the left-ventricular outflow tract [8]. Electrophysiological mechanisms of ventricular tachycardia Re-entrant ventricular arrhythmias Monomorphic ventricular tachycardia is the most common form of sustained ventricular tachycardia and usually occurs after myocardial infarction. A re-entry mechanism accounts for the majority of these ventricular tachycardias. In contrast with automatic arrhythmias, the conditions for re-entry tend to be associated with chronic rather than acute disease. Endocardial catheter mapping and intraoperative mapping have shown that these arrhythmias originate within or at the border zone of the diseased myocardium. The size of the re-entrant circuit may be large, especially in patients with a left-ventricular 951 TETC32 12/2/05 14:26 Page 952 952 Chapter 32 A 500 ms B C 200 ms 100 m/s I 1000 ms 25 m/s RF I II III III aVR V1 * aVL aVF V6 Diastolic potential V1 V2 ABL V3 V4 V5 V6 RVA Figure 32.3 ECG recording in a patient with previous anterior myocardial infarction and recurrent sustained ventricular tachycardia. (A) Catheter mapping and subsequent catheter ablation were performed. (B) Leads I, III, V1 and V6, as well as intracardiac signals from the right ventricular apex (RVA) and the ablation catheter at the successful ablation site (ABL) anteroseptal at the leftventricular base are displayed. Note: the fragmented diastolic potential at the successful ablation site (for further details, see text), where the ventricular tachycardia terminated a few seconds after starting radiofrequency (RF) ablation (C). aneurysm, or may be confined to a small area. Re-entry requires a series of conditions to be satisfied for its occurrence: (1) two potentially conducting pathways or more; (2) unidirectional block must occur in one pathway; (3) an activation wavefront that travels around that zone of unidirectional block over the alternative pathway; (4) then activation of myocardium distal to the zone of unidirectional block with delay (i.e. with slow conduction), so allowing (5) the activation wavefront to invade the zone of block retrogradely and re-excite the tissue where the activation wavefront originated. For re-entry to occur, the impulse that is conducting around the re-entrant circuit must always find excitable tissue in the direction in which it is propagating. This constellation frequently occurs in the context of myocardial scarring. An understanding of these electrophysiological phenomena is critical to the diagnosis and successful ablation of re-entrant ventricular arrhythmias. Initiation and termination of ventricular tachycardia by pacing stimuli, the demonstration of electrical activity bridging diastole and a variety of other clinically used techniques are consistent with a re-entry mechanism. Entrainment by pacing is considered the most reliable clinical method to demonstrate the presence of a re-entry mechanism. The areas of slow conduction have been shown to be desirable targets of ablation. Zones of slowly conducting myocardium may be identified during endocardial catheter mapping by fractionated and/or mid-diastolic electrograms (Fig. 32.3), continuous electrical activity or a long delay between a stimulus artefact and the resulting QRS complex. However, not all areas of slow conduction participate in the re-entry circuit, i.e. ‘dead end’ or ‘bystander’ pathways may exist. Therefore, for successful ablation, localization procedures have to provide evidence that a mapping site is actually within the re-entry circuit and is critically linked to the perpetuation of the arrhythmia. Ischaemia seems to be less frequently involved in the initiation of monomorphic ventricular TETC32 12/2/05 14:26 Page 953 Ventricular Tachycardia A C T T T T T 3 P 3 P 3 P 3 P 3 P 4 4 4 4 4 0 0 0 0 0 6 2 0 V T T T T T T T T T T T T T T T T T T S 4 S 4 S 4 S 4 S 4 S 4 S 4 S 4 S 4 S 4 S 4 S 4 D3 P 3 P3 P 3 P 3 P 3 P 0 2 0 1 0 1 0 1 0 1 1 1 3 3 3 3 3 3 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 VT VT Rx 1/Seq 2 1500 V S 1160 VT VT Rx 1/Seq 2 B I II III aVR aVL aVF V1 V2 V3 350 V4 V5 V6 Figure 32.4 (A) Episode of ventricular tachycardia (cycle length ∼ 400 ms) detected and terminated by an implantable cardioverterdefibrillator in a patient with a remote inferior myocardial infarction who experienced recurrent VT episodes. (B) Twelve-lead ECG of the VT in the same patient. (C) Posterior view of an electroanatomic voltage map (Carto) of the left ventricle. Electroanatomic mapping can be used to define isthmus boundaries and thus guide successful ablation. Colour range represents voltage amplitude. Grey denotes dense scar tissue. A linear ablation lesion was placed from the mitral annulus to the edge of the scar tissue to prohibit mitral ‘isthmus’ re-entrant tachycardias (around the mitral valve and/or around the posterior scar). tachycardia. If a VT is not inducible or haemodynamically not tolerated during an ablation procedure, electroanatomical mapping systems can be used to locate critical isthmus regions, such as the mitral isthmus (Fig. 32.4) and to guide successful ablation. Occasionally, ischaemia has been found to precede the onset of a monomorphic ventricular tachycardia. More commonly, however, acute ischaemia triggers the occurrence of polymorphic ventricular tachycardia, which may degenerate to ventricular fibrillation rather than a sustained monomorphic ventricular tachycardia. The underlying mechanisms of ventricular tachycardia in dilated cardiomyopathy are less well understood than they are in coronary artery disease. As heart failure is not a specific disease but a syndrome, there are no specific anatomical or pathological changes in failing hearts. The occurrence of ventricular tachycardia in heart failure is a result of a complex interplay between a pathological substrate and numerous environmental triggers and facilitators evoked by left-ventricular dys- function and medical therapy. The non-specific cardiac changes include diffuse interstitial fibrosis, myofibrillar degeneration and myocyte hypertrophy. All known nonspecific alterations result in disparity of electrophysiological properties within the myocardium, which provide an appropriate abnormal substrate for arrhythmogenesis. Also, a tachyarrhythmia itself may cause reversible heart failure, including myocardial changes such as dilatation and hypertrophy. Incessant or intermittent tachyarrhythmias present for months to years are known to cause reversible cardiomyopathy in experimental heart failure models as well as in patients (‘tachycardiomyopathy’). Noteworthy in the presence of heart failure due to idiopathic dilated cardiomyopathy, re-entry in the His–Purkinje system (bundle branch re-entry, Fig. 32.5) accounts for a substantial number of monomorphic ventricular tachycardias. The re-entry wavefront proceeds down one bundle branch (mostly the right bundle branch), and up the contralateral bundle. This creates a QRS complex that has a LBBB contour and a normal 953 TETC32 12/2/05 14:26 Page 954 954 Chapter 32 500 ms A B 200 ms I I II II III V1 V6 aVR aVL RA aVF V1 RBB proximal V2 V3 RBB distal V4 V5 RVA V6 Figure 32.5 ECG recording in a patient with dilated cardiomyopathy and recurrent sustained ventricular tachycardia. (A) A sustained bundle branch re-entry tachycardia with a LBBB morphology is displayed. Intracardiac signals (B) reveal ventriculo-atrial dissociation (RA, right atrial catheter; RVA, right ventricular apex) and activation of the right bundle branch (RBB) from proximal (RBB prox) to distal (RBB dis). The tachycardia was successfully ablated at the distal right bundle branch using radiofrequency current. or leftward frontal plane axis. Its significance lies in the fact that it can be easily cured by catheter ablation of the right bundle branch. Automatic ventricular arrhythmias Abnormal automaticity accounts for a minority of ventricular tachycardias. Automatic ventricular tachycardia tends to be associated with conditions such as acute myocardial infarction, hypoxaemia, electrolyte abnormalities and a high adrenergic tone. Automatic ventricular tachycardias that occur during the first 24–48 h after an acute myocardial infarction are a major cause of sudden cardiac death. They are probably related to the residual ischaemia seen acutely in the zone of infarction. Once the infarction heals, the substrate for these arrhythmias disappears (but the one for re-entry evolves). Because automatic arrhythmias generally occur secondarily to metabolic abnormalities, treatment should be aimed at identifying and reversing the underlying cause whenever possible. Triggered activity Although ventricular tachycardias based on triggered activity are uncommon, two distinct clinical syndromes involving triggered activity have been identified: pauseand catecholamine-dependent arrhythmias. In each syndrome, patients develop polymorphic ventricular tachycardia. These arrhythmias tend to occur in relatively short bursts that may be accompanied by lightheadedness or syncope, but may also degenerate into ventricular fibrillation and cause sudden death. Pause-dependent triggered activity is caused by afterdepolarizations that occur during phase 3 of the action potential (early afterdepolarizations). If these afterdepolarizations reach the threshold potential of the cardiac cell, another action potential can be generated. Pausedependent triggered activity may be related to congenital ion-channel abnormalities (long QT syndrome, see p. 961) and/or to specific conditions (hypokalaemia and hypomagnesaemia), and/or the use of non-cardiovascular or cardiovascular drugs (e.g. class IA or class III antiarrhythmic agents, i.e. acquired QT syndrome) that prolong repolarization. Individuals who develop ventricular arrhythmias (i.e. torsade de pointes) [9] in the presence of these conditions have a reduced repolarization reserve. Torsades de pointes (Fig. 32.6) is a rapid, irregular nonsustained polymorphic ventricular tachycardia that appears to twist around the isoelectric line and may TETC32 12/2/05 14:26 Page 955 Ventricular Tachycardia 10 mm/mV 25 mm/s Filter 25 Hz I II III aVR aVL aVF V1 V2 V3 V4 V5 V6 Figure 32.6 Recurrent episodes of torsade de pointes in a patient with long QT syndrome. degenerate into ventricular fibrillation. The ECG, while in sinus rhythm, usually shows prolongation of the QT interval (see long QT syndrome, p. 961). In addition, distortion of the T wave and often distinct U waves may occur. The longer the previous cycle length, the more exaggerated the TU wave aberration of the following complex, hence the condition is ‘pause-dependent’. The treatment of pause-dependent triggered activity is aimed at reducing the prolonged repolarization. Drugs that prolong the QT interval should be discontinued and avoided. Electrolyte abnormalities should be rapidly corrected. Intravenous magnesium sulphate ameliorates these arrhythmias. In addition, pauses can be eliminated by either atrial or ventricular pacing, or by beginning an isoproterenol infusion. Catecholamine-dependent triggered activity is caused by afterdepolarizations that occur during phase 4 of the cardiac action potential (delayed afterdepolarizations). They occur in the setting of congenital ion-channel abnormalities, digitalis toxicity or cardiac ischaemia. Catecholamine-dependent triggered activity generally is not dependent on pauses. Instead, these arrhythmias may arise in conditions of high sympathetic tone. Thus, patients experience ventricular tachycardia (manifested by syncope or cardiac arrest) during times of exercise or of emotional stress. Ventricular tachycardia clinical presentation The clinical presentation of ventricular tachycardia depends on the haemodynamic consequences it produces. These depend partly on ventricular tachycardia rate, the degree of myocardial dysfunction, the circumstances and suddenness of initiation, and autonomic factors. Physical examination in a patient presenting with ventricular tachycardia often indicates haemodynamic distress (low blood pressure, heart failure or cardiogenic shock). When cardiac output and blood pressure are 955 TETC32 12/2/05 14:26 Page 956 956 Chapter 32 maintained and/or when the ventricular tachycardias are short-lived, the arrhythmia may present as palpitations, breathlessness or chest pain. Sometimes, especially in patients without structural heart disease, no symptoms are reported during ventricular tachycardia. The rate of ventricular tachycardia is a major factor in determining clinical symptoms. Among 1130 patients with ventricular tachycardia, the average ventricular tachycardia rate was 163 b.p.m. in asymptomatic patients, 170 b.p.m. in patients who had lightheadedness, 191 b.p.m. in patients presenting with presyncope and 224 b.p.m. in those with syncope [10]. Persistent, slow (< 150 b.p.m.) ventricular tachycardia may lead to dyspnoea, pulmonary congestion and oedema. Patients with heart failure were more likely to present with syncope regardless of ventricular tachycardia rate. Rapid and/or persistent ventricular tachycardia, impaired left-ventricular function and atrioventricular dissociation contribute to haemodynamic collapse, which may result in presyncope, syncope or sudden death. Syncope is the single most important clinical event for grading sudden cardiac death risk in heart failure [11]. Ventricular tachycardia was found to be the cause of syncope in 35% of these patients [12]. Patients with heart failure and unexplained syncope have a 1-year sudden death rate of up to 45% [12]. The frequency and complexity of ventricular tachycardia parallel the severity of ventricular dysfunction. In total, 15–20% of patients with NYHA class I–II heart failure have non-sustained ventricular tachycardia compared with 50–70% of patients with class IV heart failure. Sustained polymorphic ventricular tachycardia is less stable than monomorphic ventricular tachycardia. It is usually rapid and often degenerates into ventricular fibrillation. Sustained monomorphic ventricular tachycardia may be haemodynamically tolerated, but may also precipitate ventricular fibrillation or may cause syncope before terminating spontaneously. Patients presenting with haemodynamically tolerated ventricular tachycardia have a lower risk of sudden cardiac death than patients whose initial episode causes cardiac arrest, but the risk is still substantial. Therapy of ventricular tachycardias in patients with structural heart disease The spectrum of therapies for ventricular tachycardias includes drug therapy, device implantation and surgical or catheter ablation interventional techniques. The management challenge is to deal with both the ventricular tachycardia that is the presenting symptom and the pervading sudden cardiac death risk that may be the consequence of the arrhythmogenic substrate. Device and drug therapy of ventricular tachyarrhythmias in patients with structural heart disease When ventricular tachycardia is the consequence of structural cardiac disease, persistence or evolution of an arrhythmogenic substrate, even after successful treatment of a presenting ventricular tachycardia, militates against any curative therapy. For a long time, therapy of ventricular tachycardia was dominated by drug therapy or anti-tachycardic surgery. However, nowadays the implantable cardioverter-defibrillator (ICD) is the best available therapy to prevent sudden cardiac death from ventricular tachycardia. In clinical use since 1980, the ICD is a self-contained device that is capable of identifying ventricular tachycardia and ventricular fibrillation and automatically terminating these arrhythmias by anti-tachycardic pacing or delivering a shock, usually about 35 J, directly to the heart. Ischaemic heart disease and idiopathic dilated cardiomyopathy Implantable cardioverter-defibrillator trials for secondary prevention of sudden cardiac death The antiarrhythmics vs. implantable defibrillator (AVID) trial [13] was the first large-scale randomized study that compared ICD therapy with antiarrhythmic drug treatment in patients with documented symptomatic ventricular tachycardia (55%) or ventricular fibrillation (45%). Patients with ventricular tachycardia also had either syncope or other serious cardiac symptoms, along with a left-ventricular ejection fraction of < 40%; 81% of these patients had coronary artery disease. In total, 1016 patients with documented ventricular tachycardia were randomized to ICD or antiarrhythmic drug therapy, almost exclusively with amiodarone. Mortality in the group treated with antiarrhythmic drugs was 17.7%, 25.3% and 35.9% after 1, 2 and 3 years respectively. The total death rate was significantly reduced by 39% in the TETC32 12/2/05 14:26 Page 957 Reduction with ICD therapy (%) Ventricular Tachycardia All cases mortality Arrhythmic death 80 70 60 50 40 30 20 10 0 AVID CASH CIDS Figure 32.7 Relative risk reduction of total death rate by ICD implantation in secondary prevention trials (for details, see text). ICD group after 1 year and by 27% and 31% after 2 and 3 years respectively. The results of AVID were consistent among all prespecified subgroups: coronary artery disease vs. other diseases, ventricular fibrillation vs. ventricular tachycardia, all age groups, and all ejection fractions. There was a small trend towards less benefit in patients with an ejection fraction above 35%. The Canadian Implantable Defibrillator Study (CIDS) [14] and the Cardiac Arrest Study Hamburg (CASH) [15] recruited similar patient cohorts as AVID (Fig. 32.7). CIDS [14] randomized 659 patients with symptomatic ventricular tachycardia, aborted sudden death or syncope in the presence of inducible ventricular tachycardia to ICD treatment or empirical amiodarone. Two-year mortality in the drug arm was about 22%. There was a reduction of total death rate by ICD implantation (risk reduction 19.6% at 3 years) but this did not reach statistical significance. In CASH [15] a total of 346 patients with a history of cardiac arrest were randomized to ICD or treatment with metoprolol, amiodarone or propafenone. After inclusion of 230 patients that were randomly assigned to propafenone, amiodarone, metoprolol or the implantable defibrillator, the propafenone arm was stopped because of excess mortality compared with the ICD group [15]. This study demonstrated a 37% survival benefit of patients receiving ICDs in comparison with metoprolol or amiodarone at 2 years. Two-year mortality in these arms was 19.6%. Noteworthy, the ejection fraction of the patients in CASH (0.46) was much higher than in AVID (0.32) or CIDS (0.34). In CASH primary ventricular fibrillation patients were also included. Data from AVID, CIDS and CASH (only amiodarone and ICD arms) were merged into a meta-analysis [16]. This analysis showed a significant reduction in death from any cause, with the ICD having a mean hazard ratio of 0.72. This 28% reduction in the relative risk of death with the ICD was largely the result of the reduction in arrhythmic death. Survival was extended by a mean of 4.4 months by the ICD over a follow-up period of 6 years. Patients with left-ventricular ejection fraction of ≤ 35% had a significantly higher benefit from ICD therapy than those with a better-preserved left-ventricular function. This was also found in a post hoc analysis of CIDS [17]. This analysis showed that three clinical risk factors were predictors of death and benefited from the ICD: age ≥ 70 years, left-ventricular ejection fraction ≤ 35% and New York Heart Association class III or IV. In contrast with patients with coronary artery disease, risk stratification in patients with idiopathic dilated cardiomyopathy is much more difficult. These patients are under-represented in all ICD studies. In AVID, CASH and CIDS only 15%, 11% and 10%, respectively, of all patients had idiopathic dilated cardiomyopathy. All of these studies showed a reduction of total mortality in patients with non-ischaemic dilated cardiomyopathy of 20–40% compared with conventional therapy [13–15]. However, the confidence intervals for patients with non-ischaemic dilated cardiomyopathy was much wider than for patients with coronary artery disease. In the meta-analysis of these three studies, only 225 out of 1832 patients had non-ischaemic cardiomyopathy [16]. These patients had a hazard ratio for reduction of total mortality of 0.78, which was very similar to the total cohort (0.72). However, the 95% confidence intervals for these patients ranged from 0.45 to 1.37. The significance of syncope in dilated cardiomyopathy without documented ventricular tachycardia is still unclear. A non-randomized study showed similar event rates of appropriate ICD discharges in patients who received an ICD because of syncope, and patients who received a defibrillator after aborted sudden death or episodes of ventricular tachycardia or ventricular fibrillation [18]. Another study showed significantly lower event rates in a series of consecutive patients treated with an ICD than in conventionally treated patients [19]. Hence, it seems reasonable to treat patients with non-ischaemic dilated cardiomyopathy and syncope similar to those after aborted sudden cardiac death if other causes of syncope are excluded. Implantable cardioverter-defibrillator trials for primary prevention of sudden cardiac death (Fig. 32.8) The Multicenter Automatic Defibrillator Implantation Trial (MADIT) [20] was the first study that showed a benefit of implanting an ICD in patients with coronary heart disease and left-ventricular dysfunction, whereas the CABG-Patch trial [21] (ICD therapy vs. no specific antiarrhythmic therapy—the other studies compared 957 TETC32 12/2/05 14:26 Page 958 Chapter 32 Reduction with ICD therapy (%) 958 All cases mortality Arrhythmic death 80 70 60 50 40 30 20 10 0 MADIT 1 MUSTT MADIT 2 SCD-HEFT Figure 32.8 Relative risk reduction of total death rate by I CD implantation in primary prevention trials (for details, see text). the ICD with antiarrhythmic drugs—for the primary prophylaxis of sudden cardiac death) in patients with impaired left-ventricular function scheduled for elective bypass surgery demonstrated no benefit. MADIT [22] enrolled patients after myocardial infarction (in 75% of the patients, the interval between infarction and enrolment was more than 6 months) with an ejection fraction below 0.36, non-sustained ventricular tachycardia and inducible ventricular tachycardia (not suppressible by a class I drug). During an average follow-up of 27 months, the risk of death was reduced by 54% in the ICD arm. MADIT II [23] was designed to investigate whether the ICD would be effective in the prevention of all-cause death in patients after myocardial infarction, with a low ejection fraction (≤ 30%) as the only inclusion criterion. A randomization ratio of 3:2 to receive an ICD or conventional therapy was selected. After inclusion of 1232 patients, the trial was terminated because of a significant (31%) reduction in all-cause death in patients assigned to ICD therapy. In a post hoc analysis, Moss and colleagues [22] found that patients with an ejection fraction of less than 26% had a far greater benefit from ICD implantation than patients with an ejection fraction of between 26% and 35% [24]. Later they identified three independent risk factors: ejection fraction < 26%, QRS duration ≥ 120 ms and a history of heart failure treatment [25]. The benefit from ICD treatment increased with the number of risk predictors. Thus, in patients with chronic coronary heart disease, the magnitude of the survival benefit from the ICD is directly related to the severity of cardiac dysfunction and its associated mortality risk. The same was found in a post hoc analysis of the Multicenter Unsustained Tachycardia Trial (MUSTT) [26]. The combination of an ejection fraction of ≤ 30% and an abnormal signal-averaged ECG identified a subgroup of particularly high risk, constituting 21% of the total study population. In contrast with MADIT and MADIT II, where 85% of the patients were included > 6 months post myocardial infarction, the yet unpublished DINAMIT study, which included patients within the first 40 days after a myocardial infarction, failed to demonstrate a benefit from prophylactic ICD implantation, despite the fact that most patients had a large anterior infarction and leftventricular ejection fraction was low (average 28%) (for comment, see ref. no. 27). Very recently, the first results of SCD-Heft trial have been presented. This trial determined if amiodarone or an ICD reduces all-cause mortality compared with placebo in patients with either ischaemic or non-ischaemic NYHA class II and III heart failure and an ejection fraction of < 35%. The ICD decreased mortality by 23%, whereas amiodarone, when used as a primary preventative agent, did not improve survival. Two other prospective studies in patients with non-ischaemic cardiomyopathy without prior arrhythmias and one in patients with asymptomatic non-sustained ventricular tachycardia have been reported. The Cardiomyopathy Trial (CAT) [28] was a pilot study in patients with a recently diagnosed (< 9 months) non-ischaemic dilated cardiomyopathy (EF < 30%) that included 102 patients. Patients were randomized to ICD therapy or no antiarrhythmic drug therapy. The primary end-point was total mortality after 2 years. In contrast with the investigators’ expectations, the total mortality after 2 years was only 8–9% in both groups. The study was terminated, as more than 1300 patients would have been needed to demonstrate a significant difference between the two groups. In the first 2 years after inclusion into the study, there was not a single case of sudden death in the control group. In the ICD group, there were 11 patients with a ventricular tachycardia faster than 200 b.p.m. All ventricular tachycardias were terminated by the ICD. Nevertheless, after 5 years only 50% of those patients with appropriate ICD discharges survived, in contrast with 85% of the patients without appropriate ICD discharges. This finding is in analogy to the finding of an association between appropriate ICD discharges and death from progressive heart failure [29]. The findings of the Defibrillators in Nonischemic Cardiomyopathy Treatment Evaluation (DEFINITE) [30,31] trial are in contrast with the results of the CAT trial. DEFINITE was the first large-scale trial investigating the use of ICD for the primary prevention of sudden cardiac death in patients with non-ischaemic dilated cardiomyopathy. It enrolled a total of 458 patients with non-ischaemic dilated cardiomyopathy, left-ventricular dysfunction (ejection fraction < 35%), NYHA class I–III heart failure and spontaneous ventricular tachycardia (premature ventricular complexes or non-sustained ventricular tachycardia). TETC32 12/2/05 14:26 Page 959 Ventricular Tachycardia Patients with unexplained syncope within 6 months, prior cardiac arrest or ventricular tachycardia of > 15 beats at a rate of > 120 b.p.m. or those on amiodarone treatment for ventricular tachycardia were excluded from the study. Patients were randomized to drug therapy with beta-blockers and ACE inhibitors (if tolerated) or drug therapy plus an ICD. The study’s primary endpoint was total mortality; the secondary end-point was arrhythmic death. During a mean follow-up of 26 months, total mortality observed in the ICD group was 8.1%, a result that did not reach statistical significance when compared with control subjects (although there was a clear trend toward a benefit). The absolute mortality benefit in the ICD group was 5.7% at 2 years and the relative risk reduction was 34%. ICDs were associated with a significantly lower rate of arrhythmic death, the study’s secondary end-point. Use of an ICD was associated with a 74% relative reduction in arrhythmic death (P = 0.01). Subgroup analyses uncovered that patients with class III heart failure who received an ICD had a 67% relative risk reduction in all-cause mortality compared with those who received drug therapy alone (P = 0.009). As ICD therapy has been shown to be beneficial in patients with impaired left-ventricular function and nonsustained ventricular tachycardia in the MADIT and the MUSTT trials [22,26], the hypothesis that ICD therapy would be superior to antiarrhythmic drug therapy also in patients with non-ischaemic dilated cardiomyopathy and non-sustained ventricular tachycardia was tested in the AMIOVIRT study [32]. Patients (n = 103) with nonischaemic dilated cardiomyopathy, left-ventricular ejection fraction of < 35% and asymptomatic non-sustained ventricular tachycardia were randomized to receive either amiodarone or an ICD. The primary end-point was total mortality. The study was stopped because of the unexpectedly low total mortality in both arms. The percent of patients surviving at 1 year (90% vs. 96%) and three years (88% vs. 87%) in the amiodarone and ICD groups, respectively, was not different. As there was no true placebo group in this study, it cannot be clarified whether non-sustained ventricular tachycardia is useful as a risk predictor in non-ischaemic dilated cardiomyopathy or whether amiodarone is highly efficient in this patient cohort. The latter had already been suggested retrospectively by the CHF-STAT trial, when amiodarone proved more effective in non-ischaemic vs. ischaemic patients [33]. However, in a prospective registry including 343 patients with idiopathic dilated cardiomyopathy, only reduced left-ventricular EF and lack of beta-blocker therapy were predictors of an increased arrhythmic risk [34]. Signal-averaged ECG, QTc dispersion, heart rate variability, baroreflex sensitivity and microvolt T-wave alternans did not predict arrhythmia risk, and non- sustained ventricular tachycardia on Holter was associated only with a trend towards higher arrhythmia risk. In contrast with these findings, patients with nonsustained ventricular tachycardia in the CAT trial had a markedly increased total mortality rate with only 63% surviving after 6 years compared with 77% of the patients without non-sustained ventricular tachycardia. However, in CAT, even in this subgroup, there was no benefit from ICD implantation. In patients with nonischaemic dilated cardiomyopathy, non-sustained ventricular tachycardia seems to be more a marker for increased total mortality than for a high arrhythmic risk. Catheter ablation or surgical treatment of ventricular tachyarrhythmias Catheter ablation might be an adjunctive but rarely curative option for a highly select group of patients with refractory or incessant ventricular tachycardia (i.e. patients with multiple ICD discharges due to ventricular tachycardia—Fig. 32.3). Catheter ablation has been successfully applied in ventricular tachycardia that is caused by ischaemic heart disease. Results with computerized mapping systems and mapping techniques using noncontact mapping systems that do not require sustained ventricular tachycardia during the ablation procedure have demonstrated promising results in ventricular tachycardia ablation. Surgical techniques for treatment of ventricular tachycardia may be effective in ICD carriers with sustained monomorphic ventricular tachycardia resulting from coronary artery disease, especially when a discrete left-ventricular aneurysm and inducible monomorphic ventricular tachycardia are present [35]. In selected patients, anti-tachycardia operations can be carried out with an acceptable mortality and a relatively high long-term survival rate. However, these procedures cannot be expected to alter the natural history of the underlying heart disease. Bundle branch re-entry ventricular tachycardia, which may be relatively commoner in idiopathic dilated cardiomyopathy, is particularly amenable to catheter ablation (see above). Other cardiomyopathic conditions Arrhythmogenic right ventricular cardiomyopathy Arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVC) was first described in 1982 [36] and since 959 TETC32 12/2/05 14:26 Page 960 960 Chapter 32 then has been diagnosed with increased frequency. ARVC is a primary myocardial disorder with a genetic background. In recent years, the disease has been recognized as a major cause of ventricular arrhythmias and sudden death, particular in young patients and athletes with apparently normal hearts. ARVC is characterized by localized or diffuse atrophy of predominantly rightventricular myocardium, with subsequent replacement with fatty and fibrous tissue, and usually manifests with ventricular tachycardia and/or sudden death, frequently before structural abnormalities become apparent [36– 38]. Diagnostic criteria of ARVC were proposed by an international study group [39] and include major and minor criteria in different categories. Eight chromosomal loci for autosomal dominant forms of ARVC and two loci for autosomal recessive inheritance (one of which is Naxos disease) have been reported. In Naxos disease, a syndromic variant of ARVC with palmoplantar keratosis and woolly hair, a mutation in the gene encoding the cytoskeletal protein plakoglobin was identified. Several years later, a mutation in the desmoplakin gene, another protein involved in cell-to-cell junctions (adherens junctions and desmosomes), was identified in a classical form of ARVC (ARVC-8), with frequent left-ventricular involvement. In a rare and rather atypical subgroup of ARVC (ARVC-2) with minor right ventricular abnormalities and polymorphic ventricular arrhythmias, a mutation in the gene encoding the cardiac ryanodine receptor (RyR2) was identified. In ARVC, episodes of ventricular tachycardia are frequently well tolerated, mainly due to the preserved left-ventricular function. Antiarrhythmic treatment of ARVC includes drug therapy, catheter ablation and ICD implantation. The available but limited data on risk stratification indicate that patients with severe right ventricular dysfunction, left-ventricular involvement, a history of syncope or cardiac arrest, family history of sudden cardiac death, inducible ventricular tachycardia/ventricular fibrillation and ECG abnormalities (epsilon potential, late potential) are more prone to life-threatening ventricular tachycardia and sudden death. In patients with ARVC and low risk of sudden death, antiarrhythmic drug therapy is an alternative option. Low-risk cohorts include patients with localized right-ventricular disease and monomorphic ventricular tachycardia suppressed by antiarrhythmic drugs. Despite high efficacy rates of radiofrequency catheter ablation in abolishing regional sites of ventricular tachycardia, there is a high recurrence rate due to new ventricular tachycardia morphologies and origins. Main indications for catheter ablation in ARVC include monomorphic ventricular tachycardia in localized rightventricular abnormalities and incessant or frequent ven- tricular tachycardia not suppressed by antiarrhythmic treatment. Recent studies [40,41] in high-risk patients with ARVC after resuscitated cardiac arrest, life-threatening ventricular tachycardia or drug-refractory ventricular tachycardia demonstrated the high efficacy of ICD implantation in the prevention of sudden death. The estimated survival benefit of ICD therapy was 21%, 32% and 36% after 1, 3 and 5 years, respectively, of follow-up. The role of ICD therapy for primary prevention of sudden death in ARVC remains unclear to date because only very preliminary data are available. Patients with welltolerated and non-life-threatening ventricular tachycardia are usually treated empirically with antiarrhythmic drugs, including amiodarone, sotalol, beta-blockers, flecainide and propafenone, alone or in combination. ICDs are usually reserved for patients with life-threatening ventricular tachycardia, in whom drug therapy is either ineffective or undesirable. Wichter and colleagues [41] found that, in a series of 60 patients in a single centre during a mean follow-up of 80 ± 43 months, event-free rate after 5 years was only 26% for ventricular tachycardias and 59% for potentially fatal ventricular tachycardias with a rate > 240 b.p.m. Extensive rightventricular dysfunction was identified as a predictor for appropriate ICD discharges. Hypertrophic cardiomyopathy Hypertrophic cardiomyopathy (HCM) is an inherited myocardial disorder with an autosomal dominant trait and is caused by mutations in one of 10 genes known so far, each encoding for protein components of the cardiac sarcomer. There is broad heterogeneity not only concerning disease-causing genetic mutations, but also in terms of phenotypic expression, treatment and prognosis. Patients with hypertrophic cardiomyopathy often present with ventricular ectopy or non-sustained ventricular tachycardias that are associated with a high risk of sudden cardiac death. Symptoms of HCM range from dyspnoea and angina pectoris to palpitations, dizziness and syncope [42]. Treatment of symptomatic HCM patients includes drugs (verapamil, beta-blockers or disopyramide) or nonpharmacological options (septal myectomy, DDD pacing, alcohol septal ablation) in those with obstructive HCM [43]. These treatment options are targeted to reduce symptoms and improve quality of life, but have not been shown to have an impact on survival. Sudden cardiac death may occur without warning signs or symptoms, as the initial disease manifestation, and may be triggered by vigorous exercise or competitive sports activity. The highest risk for sudden cardiac death has been associated with prior cardiac arrest or spontaneous sustained ventricular tachycardia/ventricular TETC32 12/2/05 14:26 Page 961 Ventricular Tachycardia fibrillation. In such patients, the implantation of an ICD is strongly recommended for secondary prevention of sudden death. In a multicentre retrospective study in high-risk HCM patients, appropriate ICD interventions occurred in 25% of patients after a follow-up period of only 3 years. Potentially life-saving ICD therapies were reported at a rate of 11% per year in patients receiving the ICD for secondary prevention (aborted sudden death or sustained ventricular tachycardia/ventricular fibrillation), compared with a rate of 5% per year in the primary prevention cohort (based solely on non-invasive risk factors) [44]. In the setting of primary prevention, major risk factors for sudden death in HCM include a high-risk mutant gene, a family history of premature sudden death, unexplained syncope, abnormal exercise blood pressure, non-sustained ventricular tachycardia (Holter), and severe left-ventricular hypertrophy (≥ 30 mm). In individual patients, atrial fibrillation, myocardial ischaemia, leftventricular outflow-tract obstruction and vigorous physical exertion or competitive sports may be additional risk factors [45,46]. ICD implantation is considered the most effective and reliable treatment option and has been recommended in HCM patients at high risk of sudden death [44–46]. HCM patients without risk factors are at low risk of sudden death and should be reassured and followed clinically. Little or no restriction is necessary with regard to employment and recreational activities but patients should be excluded from strenuous exercise and competitive sports. Table 32.1 Ventricular tachycardias in patients with primary electrical disease and inherited myocardial diseases ‘Primary electrical disorders’, in which an organic heart disease is not detectable Long QT syndrome (LQTS) Short QT syndrome (SQTS) Catecholaminergic polymorphic ventricular tachycardia (CPVT) Idiopathic right-ventricular outflow tract tachycardia (RVOT-VT) Idiopathic left-ventricular tachycardias (ILVT) Idiopathic ventricular fibrillation (IVF) Brugada syndrome ‘Arrhythmogenic cardiomyopathies’, in which an inherited myocardial disease may primarily manifest with ventricular tachyarrhythmias Arrhythmogenic right ventricular cardiomyopathy (ARVC) Hypertrophic cardiomyopathy (HCM) Dilated cardiomyopathy (DCM) precipitating factors (e.g. exercise), site of origin (i.e. left or right ventricle), by response to antiarrhythmic drugs (e.g. adenosine or verapamil) or on the basis of an underlying organic heart disease (primary electrical disorder vs. inherited myocardial disease—Table 32.1). Ventricular tachycardia in patients without structural heart disease but not currently amenable to curative therapies Ventricular tachycardia in patients without structural heart disease: ‘idiopathic’ ventricular tachycardia ‘Idiopathic ventricular tachycardia’ is a non-specific term that represents a heterogeneous group of arrhythmias. Awareness of this entity has existed since it was first described by Gallavardin [47] in 1922. Patients can be completely asymptomatic or have transient symptoms including palpitations, dizziness or presyncope, but these arrhythmias, with the exception of rapid polymorphic ventricular tachycardia or idiopathic ventricular fibrillation occurring in the setting of inherited arrhythmic syndromes, are rarely life threatening. The underlying mechanisms include re-entry, triggered activity and catecholamine-mediated automaticity. Idiopathic ventricular tachycardia can be categorized according to the clinical presentation (non-sustained vs. sustained), Long QT syndrome (LQTS) is characterized by a prolonged QT interval in the surface ECG, recurrent syncope or sudden death resulting from torsade de pointes (Fig. 32.6) [48–51]. The incidence of LQTS has been estimated as 1 in 7000 to 1 in 10 000 live births. More than 250 mutations in seven genes (LQTS 1–7) have been described. Mutations involve genes encoding potassium channels [LQT1, 2, 5 and 6, Jervell Lange–Nielsen ( JLN) 1 and 2], sodium channels (LQT3) and ankyrin B (LQT4), which acts as a targeting and anchoring molecule for the sodium channel. In 30–40% of all patients with LQTS, no gene defect can be found, pointing towards a large heterogeneity of gene loci. The term acquired LQTS [52,53] is reserved for a syndrome similar to the congenital form but caused by exposure to drugs that prolong the duration of the ventricular action potential or to QT prolongation secondary to bradycardia or an electrolyte 961 TETC32 12/2/05 14:26 Page 962 962 Chapter 32 imbalance. Drugs that prolong the QT interval, and thereby predispose to torsade de pointes, are listed on websites such as www.qtdrugs.org. Associations between genotype and phenotype have been investigated based on the International LQTS Registry, which was started in 1979. Moss and colleagues [54] identified a gene-specific phenotype in the repolarization pattern of the surface ECG. Patients with LQT1 show a broad and prolonged T wave, whereas LQT2 patients have a notched, low-amplitude T wave. Patients of the LQT3 demonstrate a long isoelectric ST segment with a delayed, peaked narrow T wave. In LQT3, cardiac events occur more frequently at rest or during sleep, whereas they are typically related to emotion or exercise (in particular, swimming) in LQT1 and auditory stimuli in LQT2 [55,56]. Of 533 genotyped index patients (243 LQT1, 209 LQT2, 81 LQT3) and 1842 family members mortality was highest in patients with LQT3 followed by male patients with LQT1 and 2 and female patients with LQT1 and 2, but arrhythmic events occurred more frequently in LQT1 and LQT2 [57]. Priori and colleagues [55] presented a scheme for risk stratification, based on analysis of 647 patients. High risk was considered in patients with LQT1 and QTc > 500 ms1/2 and in male patients with LQT2 or LQT3 and QTc > 500 ms1/2. High-risk patients should be treated prophylactically using beta-blockers [58], although the effect is less beneficial in patients with LQT3 [59]. Beta-blocker therapy is associated with a significant reduction in the rate of cardiac events. Event rates within 5 years while on betablocker were higher in those patients who were symptomatic before starting this therapy (32%) than in those who had been asymptomatic (14%) [60]. Subgroup analysis in genotyped patients with LQT1, LQT2 and LQT3 showed that beta-blocker therapy had only minimal effects on QTc in all three genotypes. Treatment was associated with a significant reduction of events in LQT1 and LQT2 patients, whereas there was no evident effect in LQT3 [60]. In selected patients with LQT1, LQT2 and LQT5, potassium channel openers (i.e. pinacidil, nicorandil) may become a therapeutic option [61,62]. In LQT3, mexiletine may selectively suppress the mutant channel phenotype by inhibition of late openings [63,64] and lidocaine showed similar effects [65]. Priori and colleagues [66] were able to show a significant reduction of QTc prolongation in LQT3 patients carrying mutant sodium channels that were known to be influenced by mexiletine in vitro. Similar effects were reported with flecainide [67]. There is only limited information available on the role of ICD therapy in patients with LQTS. The ACC/AHA 2002 guidelines have designated the ICD for primary prevention of SCD as a class IIb indication. In clinical practice, the decision for prophylactic ICD implantation is not based on gene analysis. Usually, prophylactic ICD implantation is considered in patients with syncope despite beta-blocker therapy or in patients with syncope and with a family history of sudden death. A benefit from ICD has been suggested by retrospective analyses. Zareba and colleagues [68] compared 73 LQTS patients who were treated with ICD because of prior cardiac arrest (n = 54) or recurrent syncope despite beta-blocker therapy (n = 19) with 161 LQTS patients who had similar indications (89 cardiac arrest and 72 recurrent syncope despite beta-blocker therapy) but did not receive ICD. There was one (1.3%) death in 73 ICD patients following an average of 3 years, whereas there were 26 deaths (16%) in non-ICD patients during a mean 8-year follow-up. However, it was noted by Viskin [69] that, after exclusion of the patients who died within 1 month after inclusion and therefore likely from residuals of their first aborted sudden death, the difference between both groups was only marginal. Hence, a long-term prospective study is needed to determine the benefit of ICD therapy in LQTS. Short QT syndrome Very recently, a new syndrome associated with sudden cardiac death in otherwise healthy patients with structurally normal hearts has been described, the short QT syndrome [70,71]. The prevalence of this syndrome is unknown. Patients with the short QT syndrome (SQTS) present with a short QT interval on the 12-lead ECG, familial sudden death and palpitations, syncope or sudden cardiac arrest. Six patients from two European families were extensively tested by non-invasive and invasive methods. Mean QT intervals were 252 ± 13 ms (QTc = 287 ± 13 ms). In four patients, electrophysiological studies were performed, revealing short atrial and ventricular refractory periods in all and an increased propensity to ventricular vulnerability to fibrillation in three out of four patients [70,71]. The genetic basis has only recently been uncovered. In two families, two different missense mutations of the cardiac potassium channel HERG (KCNH2) were identified, resulting in the same amino acid change. These mutations dramatically increase the potassium current IKr, leading to heterogeneous abbreviation of action potential duration and refractoriness. The affinity of the affected channels to IKr blockers is markedly reduced [72]. Currently, ICD implantation is the only therapeutic option. First experience with ICD therapy in SQTS indicates an increased risk of inappropriate device discharge owing to atrial fibrillation and T-wave oversensing, which constitutes a significant and specific risk in patients with SQTS [73]. TETC32 12/2/05 14:26 Page 963 Ventricular Tachycardia Catecholaminergic polymorphic ventricular tachycardia Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a clinically and genetically heterogeneous disease. It is characterized by episodes of syncope or sudden death in response to physiological or emotional stress occurring in structurally normal hearts [74–79]. Documented arrhythmias include bidirectional ventricular tachycardia, polymorphic ventricular tachycardia and, in rare patients, catecholaminergic idiopathic ventricular fibrillation. CPVT was first described in a Bedouin tribe from Israel [80] but has also been identified in other populations [77,81,82]. A family history of juvenile sudden death and stress-induced syncope is present in approximately one-third of cases. Mortality is high and reaches up to 30–50% by the age of 30 years [83]. Around 40–60% of the patients with CPVT carry mutations in the cardiac ryanodine receptor gene (RyR2) [82] or in the calsequestrin 2 gene (CASQ2) [84]. Genotype–phenotype analysis showed that men are at higher risk of cardiac events (i.e. syncope) and that mutation carriers became symptomatic at a younger age [76]. Current treatment of CPVT consists of β-adrenergic blockers [76,80], antiarrhythmic drugs and/or ICD implantation, mainly based on empirical grounds or the results of serial exercise/ pharmacological testing [83]. Idiopathic ventricular fibrillation In 5–10% of survivors of cardiac arrest due to ventricular arrhythmias, no structural abnormality of the heart as the underlying cause is found. In the absence of demonstrable structural heart disease, myocardial ischaemia, drug effects, electrolyte or metabolic abnormalities and toxicity, and ventricular fibrillation and unexplained cardiac arrest is rare [85 – 88]. However, it appears to be more frequent than previously thought and accounts for approximately 6–12% of all sudden deaths (lifetime prevalence < 0.5 in 10 000), with a higher percentage in the young population below the age of 40 years. Ventricular fibrillation in patients with apparently normal hearts may represent a true ‘primary electrical disease’, but it may also be the first manifestation of a cardiomyopathy. The diagnosis of idiopathic ventricular fibrillation must therefore be made by exclusion, implying that adequate and extensive diagnostic evaluation is necessary in order to rule out subclinical structural heart disease. Idiopathic ventricular fibrillation is associated with a high mortality rate. Available data suggest an 11% rate of sudden death within 1 year of diagnosis or recurrence rates of up to 30% 5 years after an initial episode of survived cardiac arrest [85]. Therefore, effective treatment is mandatory to improve the long-term prognosis. In one report, quinidine (class IA antiarrhythmic agent) was highly effective in preventing arrhythmia re-induction during electrophysiological study [88]. ICD implantation is currently the treatment of choice in patients with idiopathic ventricular fibrillation in order to prevent sudden death from recurrent episodes of ventricular fibrillation. In selected patients, catheter ablation may be a potential new option in the treatment of idiopathic ventricular fibrillation by targeting premature ventricular beats arising from the Purkinje conducting system, which have been observed to trigger polymorphic ventricular tachycardia [89]. Brugada syndrome In 1992, Brugada and Brugada [90] reported a new clinical entity with a RBBB pattern and ST segment elevation in right precordial ECG leads (Fig. 32.11) and a high incidence of sudden cardiac death in patients with structurally normal hearts. The disease is considered as a subgroup of idiopathic ventricular fibrillation and has been referred to as Brugada syndrome. It manifests with episodes of polymorphic ventricular tachycardia, syncope, and cardiac arrest during adulthood at a mean age of 40 years but within a large age range. Because symptoms occur mostly at night, this syndrome is also assigned as ‘sudden unexpected nocturnal death syndrome’. It accounts for approximately 4–12% of sudden deaths and for 20–40% of sudden cardiac arrest in patients without structural heart disease. It dominantly occurs in males and appears to be most prevalent in South-East Asia and Japan, where the disorder is a leading cause of natural death among young men with an estimated annual mortality rate of 26–38 per 100 000 [91]. Diagnostic criteria were recently proposed and reported in a consensus document [92] and mainly rely on electrocardiographic abnormalities after exclusion of structural heart disease by detailed cardiac investigation. Before making the diagnosis of Brugada syndrome, it is mandatory to exclude myocardial ischaemia and organic heart disease, particularly affecting the right ventricle (i.e. arrhythmogenic right-ventricular cardiomyopathy) as well as extracardiac and electrolyte abnormalities. Brugada syndrome is considered a ‘channelopathy’ that belongs to the group of ‘primary electrical diseases’ of the heart. In familial Brugada syndrome (20–30%), genetic mutations identified so far refer to the alphasubunit of the cardiac sodium channel (SCN5A) [93]. Assessment of these mutations in expression systems demonstrated loss of function of the sodium channel. 963 TETC32 12/2/05 14:26 Page 964 964 Chapter 32 B A I Pulmonic valve II III aVR aVL Site of RF ablation aVF V1 C V2 V3 V4 V5 V6 Figure 32.9 Non-contact mapping of a ventricular bigeminus with LBBB inferior axis morphology originating from the right ventricular outflow tract in a highly symptomatic patient (A). The multielectrode array catheter (MEA) is part of the non-contact mapping system (EnSite 3000; Endocardial Solutions). The system permits mapping of a single complex. The MEA, which is filled with a contrast saline medium, is positioned in the right-ventricular outflow tract (RAO/LAO, right/left anterior oblique views). The system calculates electrograms from 3000 endocardial points simultaneously by reconstructing far-field signals. Non-depolarized myocardium is shown in purple in this three-dimensional isopotential map (B). The map also shows the site of earliest depolarization (white circle). At this site the extrasystoles were successfully ablated using radiofrequency ablation. The ablation catheter is located at the successful ablation site. RA, diagnostic catheter in the right atrium (C). However, mutations in the SCN5A gene have been detected in only a minority of patients, thus indicating genetic heterogeneity. In the more common sporadic disease (70–80%), mutations in the SCN5A gene are very infrequent [94]. Although, at present, genetic testing is not helpful in risk stratification and clinical decisionmaking, it is important for the expansion of pathophysiological knowledge and understanding of genotype– phenotype correlations [95]. Three types of repolarization patterns can be described in Brugada syndrome [92]. Type 1 demonstrates a coved ST segment elevation of ≥ 2 mm (0.2 mV) and negative T waves in the right precordial leads (Fig. 32.11). Type 2 is characterized by a saddleback appearance with a high take-off ST segment elevation (≥ 2 mm) followed by a gradually descending ST segment (remaining ≥ 1 mm above baseline) and positive or biphasic T waves. Type 3 has either coved or saddleback morphology, with an ST segment elevation of < 1 mm. These electrocardiographic manifestations of Brugada syndrome may be transient or concealed but can be unmasked or challenged with sodium channel blockers (i.e. ajmaline, flecainide and others) [96,97], vagotonic stimulation [98] or fever [99]. Only type 1 should be considered diagnostic. Types 2 and 3 must be considered suspicious and an ajmaline test has to be performed to uncover type 1 for the diagnosis. The diagnostic and prognostic impact of an incidental finding of Brugada-type ECG signs in asymptomatic individuals without a family history represents a controversial and currently unresolved yet growing problem in clinical decision-making. Because ventricular fibrillation is the most important and frequently first manifestation of Brugada syndrome, appropriate diagnosing and early risk stratification are vital for patient management and prevention of sudden cardiac death. Brugada and colleagues [100] identified male gender, TETC32 12/2/05 14:26 Page 965 Ventricular Tachycardia spontaneous ST segment elevation and inducible ventricular tachycardia/ventricular fibrillation as indicators of high risk. In their study population, patients with a family history of Brugada syndrome appeared not to be at increased risk when compared with those with sporadic disease. Patients with an episode of aborted sudden death were at highest risk for recurrent arrhythmic events, whereas symptomatic (i.e. syncope) and asymptomatic patients with spontaneous ST segment elevation were at moderate risk. In these patients, the result of programmed electrical stimulation appeared to be helpful in clinical decision-making. Asymptomatic patients with ST segment elevation only after challenge with sodium channel blockers were at low risk for life-threatening arrhythmias [100]. The role of programmed electrical stimulation for risk stratification has been a matter of controversial discussion. Some studies [96,101,102] failed to find a correlation between ventricular tachycardia/ventricular fibrillation recurrence and inducibility of ventricular tachyarrhythmias. Priori and colleagues [102] collected clinical data from 200 patients with Brugada syndrome and identified patients with the combined presence of a spontaneous right precordial ST segment elevation and the history of syncope at highest risk of sudden death (hazard ratio 6.4; P < 0.002). Spontaneous ST segment elevation of ≥ 2 mm without history of syncope indicated intermediate risk (hazard ratio 2.1, not significant). A history of syncope per se and the results of programmed electrical stimulation were not helpful in identifying individuals at higher risk of major arrhythmic events [102]. Very recently, Eckardt and colleagues [103] reported data on a large population of individuals with a type 1 Brugada ECG pattern. During a mean follow-up of 40 ± 50 months, 4 out of the 24 patients (17%) with aborted sudden cardiac death and 4 out of 65 (6%) with a prior syncope had a recurrent arrhythmic event, whereas only 1 out of 123 asymptomatic individuals (0.8%) had a first arrhythmic event. A previous history of aborted sudden death or syncope and the presence of a spontaneous type 1 ECG were the only significant predictors of adverse outcome. The results of programmed electrical stimulation correlated only poorly to outcome. Hence, available data on risk stratification for symptomatic and asymptomatic patients in Brugada syndrome are inconclusive, and patient management and therapeutic strategies are controversial and under constant debate and refinement. β-Adrenergic (i.e. isoproterenol) or anticholinergic agents may be helpful in restoring the balance of currents during phase 1, whereas beta-blockers and amiodarone have demo strated no clinical efficacy [104]. Reports from Belhassen and colleagues [105,106] indicate a potential efficacy of quinidine in the treat- ment of ventricular tachycardia and prevention of sudden death in Brugada syndrome [107]. However, systematic or randomized studies on the clinical efficacy of quinidine in Brugada syndrome are not available. Currently, ICD implantation is the treatment of choice in secondary and primary prevention of sudden death in high-risk patients with Brugada syndrome [105,108]. Ventricular tachycardia in patients without structural heart disease, who are amenable to curative therapies Idiopathic right-ventricular outflow-tract ventricular tachycardia This arrhythmia, which has also been termed repetitive monomorphic ventricular tachycardia, usually originates in the right ventricular outflow tract. It is usually seen in younger patients (female > male) without structural heart disease and accounts for up to 70% of idiopathic ventricular tachycardia. Although the majority of cases appear to occur sporadically rather than on a familial basis, the condition is generally considered as a ‘primary electrical disease’. It is important in the differential diagnosis of various entities, in particular mild or subclinical forms of arrhythmogenic right ventricular cardiomyopathy [109]. Most data suggest that the mechanism of RVOT-ventricular tachycardia is triggered activity due to adenylcyclase-mediated delayed afterdepolarizations [110]. They are usually exertion- or stress-related arrhythmias. They can also present as recurrent extrasystolies (Fig. 32.9) or non-sustained arrhythmias tending to occur at rest (‘repetitive monomorphic ventricular tachycardia’), or provoked only with exercise (Gallavardin’s tachycardias [47]). However, these forms may just represent different spectra of the same arrhythmia. Idiopathic RVOT-ventricular tachycardia is usually well tolerated, probably owing to the preserved ventricular function. Hence, RVOT-ventricular tachycardia has a favourable long-term prognosis compared with ventricular tachycardia in structural heart disease. It manifests as a left bundle branch block ventricular tachycardia with an inferior axis (Fig. 32.9). Pacing the heart at a rapid rate or isoproterenol infusion can often induce the arrhythmia. The arrhythmia is responsive to therapy with beta-blockers [111], sotalol [111,112] or calcium channel blockers [110,113] and can also be amenable to transcatheter ablation (Fig. 32.9) [109,114]. 965 TETC32 12/2/05 14:26 Page 966 966 Chapter 32 1s A 2s B Aortic valve I II III aVR aVL aVF V1 C V2 I V3 II V1 V4 V5 Site of RF ablation Abl V6 3s Figure 32.10 Non-contact mapping of an idiopathic left-ventricular tachycardia with RBBB left axis deviation (A). The multiple electrode array (Ensite 3000, Endocardial Solutions) was placed in the left ventricle (for details, see legend to Fig. 32.9). At the distal part of the left posterior fascicle, radiofrequency ablation almost immediately terminated the ventricular tachycardia (C), which, thereafter, was no longer inducible. Idiopathic left-ventricular tachycardias (fascicular ventricular tachycardia) This arrhythmia tends to occur in younger, predominantly male patients, without structural heart disease [115,116]. An association with exertion or stress is uncommon. The arrhythmia has a relatively narrow (0.10– 0.14 s) RBBB morphology with a rapid downstroke of S waves in the precordial leads and a left superior axis (Fig. 32.10). It is inducible with programmed stimulation. ILVT is thought to have a re-entrant basis or derives from triggered activity secondary to delayed afterdepo- larizations [117]. It arises on or near to the septum near the left posterior fascicle [118–121]. Rarely, ventricular tachycardia can arise from the left anterior fascicle [115] and thus produce an RBBB pattern with right-axis deviation. Catheter ablation (Fig. 32.10) [122] offers curative therapy and should be considered early in the management of symptomatic patients. It can be performed using pace mapping [118,123], presystolic Purkinje potential [123,124] or diastolic potential during ventricular tachycardia [120,120]. Alternatively, ILVT tends to respond to therapy with beta-blockers and calcium channel blockers [112,115]. TETC32 12/2/05 14:26 Page 967 Ventricular Tachycardia A B I aVR II aVL III aVF V1 V4 V2 V5 V3 V6 O V V Figure 32.11 Twelve-lead ECG of a resuscitated patient with Brugada syndrome. The ECG is characterized by a prominent coved ST segment elevation displaying a J wave amplitude or ST segment amplitude elevation of ≥ 0.2 mV at its peak, followed by a negative T wave, with little or no isoelectric separation (A). Patients with such an ECG may develop syncope or sudden cardiac death due to fast polymorphic ventricular tachycardia (B: for details, see text). Personal perspective During the recent decades, our understanding of the clinical problem of ventricular tachycardia has markedly changed. At the beginning of my training in the early 1970s, ventricular tachycardia was a problem that seemed to represent a common entity. It took some time to understand that the mechanisms and the prognostic implications of sustained ventricular tachycardia often were markedly different, despite similar electrocardiographic appearances. Our understanding was greatly improved by experimental and clinical–electrophysiological studies for which the introduction of programmed electrical stimulation by the late Philippe Coumel and by Hein J.J. Wellens was of paramount importance. Experimental and clinical work, often done by the same persons or at least the same groups, have fertilized each other and have contributed to the rapid expansion of electrophysiological studies, drug assessment, techniques for localization of the underlying electrophysiological substrate, antitachycardia surgery, catheter ablation and, finally, as the now established therapy of first choice in most cases, the implantable cardioverter-defibrillator pioneered by Michel Mirowski. We had to learn that not everything is re-entry but that abnormal automaticity, especially after depolarization, plays an important role, too. Seminal observations, such as the one by Dessertenne, describing torsades de pointes as a specific type of ventricular tachycardia, were followed by decades of clinical observations and experimental and pharmacological studies, which finally led to the identification of the underlying molecular genetic background of long QT syndromes. Nowadays, ventricular tachycardia, appearing under different aetiologies, can frequently be viewed as separate entities with different electrophysiological substrates, different arrhythmia mechanisms and often markedly different prognosis. The spectrum of therapy has changed with almost complete disappearance of anti-tachycardia surgery and, instead, dominance of the implantable cardioverter-defibrillator in patients at risk of sudden cardiac death. For me, the field of experimental and clinical research on ventricular tachycardia is one of the almost ideal examples of translational research ‘from bench to bedside’ and vice versa. 967 TETC32 12/2/05 14:26 Page 968 968 Chapter 32 References 1 Bayes-de-Luna A, Coumel P, Leclercq JF. Ambulatory sudden cardiac death: mechanisms of production of fatal arrhythmia on the basis of data from 157 cases. Am Heart J 1989; 117: 151–159. 2 Akhtar M, Shenasa M, Jazayeri M, Caceres J, Tchou PJ. Wide QRS complex tachycardia. Reappraisal of a common clinical problem. Ann Intern Med 1988; 109: 905 –912. 3 Baerman JM, Morady F, DiCarlo LA, Jr, De Buitleir M. Differentiation of ventricular tachycardia from supraventricular tachycardia with aberration: value of the clinical history. Ann Emerg Med 1987; 16: 40 –43. 4 Brugada P, Brugada J, Mont L, Smeets J, Andries EW. A new approach to the differential diagnosis of a regular tachycardia with a wide QRS complex. Circulation 1991; 83: 1649 –1659. 5 Kindwall KE, Brown J, Josephson ME. Electrocardiographic criteria for ventricular tachycardia in wide complex left bundle branch block morphology tachycardias. Am J Cardiol 1988; 61: 1279 –1283. 6 Wellens HJ, Bar FW, Lie KI. The value of the electrocardiogram in the differential diagnosis of a tachycardia with a widened QRS complex. Am J Med 1978; 64: 27–33. 7 Kremers MS, Black WH, Wells PJ, Solodyna M. Effect of preexisting bundle branch block on the electrocardiographic diagnosis of ventricular tachycardia. Am J Cardiol 1988; 62: 1208 –1212. 8 Kamakura S, Shimizu W, Matsuo K et al. Localization of optimal ablation site of idiopathic ventricular tachycardia from right and left-ventricular outflow tract by body surface ECG. Circulation 1998; 98: 1525 –1533. 9 Dessertenne F. La tachycardie ventriculaire à deux foyers opposés variables. Arch Mal Coeur 1966; 59: 263 –272. 10 Morady F, Shen EN, Bhandari A, Schwartz AB, Scheinman MM. Clinical symptoms in patients with sustained ventricular tachycardia. West J Med 1985; 142: 341–344. 11 Packer M. Sudden unexpected death in patients with congestive heart failure: a second frontier. Circulation 1985; 72: 681– 685. 12 Stevenson WG, Middlekauff HR, Stevenson LW, Saxon LA, Woo MA, Moser D. Significance of aborted cardiac arrest and sustained ventricular tachycardia in patients referred for treatment therapy of advanced heart failure. Am Heart J 1992; 124: 123 –130. 13 A comparison of antiarrhythmic-drug therapy with implantable defibrillators in patients resuscitated from near-fatal ventricular arrhythmias. The Antiarrhythmics versus Implantable Defibrillators (AVID) Investigators. N Engl J Med 1997; 337: 1576 –1583. 14 Connolly SJ, Gent M, Roberts RS et al. Canadian implantable defibrillator study (CIDS): a randomized trial of the implantable cardioverter defibrillator against amiodarone. Circulation 2000; 101: 1297–1302. 15 Kuck KH, Cappato R, Siebels J, Ruppel R. Randomized comparison of antiarrhythmic drug therapy with implantable defibrillators in patients resuscitated from cardiac arrest: The Cardiac Arrest Study Hamburg (CASH). Circulation 2000; 102: 748 –754. 16 Connolly SJ, Hallstrom AP, Cappato R et al. Meta-analysis of the implantable cardioverter defibrillator secondary prevention trials. AVID, CASH and CIDS studies. Antiarrhythmics vs Implantable Defibrillator study. Cardiac Arrest Study Hamburg. Canadian Implantable Defibrillator Study. Eur Heart J 2000; 21: 2071–2078. 17 Sheldon R, Connolly S, Krahn A, Roberts R, Gent M, Gardner M. Identification of patients most likely to benefit from implantable cardioverter-defibrillator therapy: the Canadian Implantable Defibrillator Study. Circulation 2000; 101: 1660 –1664. 18 Knight BP, Goyal R, Pelosi F et al. Outcome of patients with nonischemic dilated cardiomyopathy and unexplained syncope treated with an implantable defibrillator. J Am Coll Cardiol 1999; 33: 1964–1970. 19 Fonarow GC, Feliciano Z, Boyle NG et al. Improved survival in patients with nonischemic advanced heart failure and syncope treated with an implantable cardioverter-defibrillator. Am J Cardiol 2000; 85: 981–985. 20 Moss AJ, Hall WJ, Cannom DS et al. Improved survival with an implanted defibrillator in patients with coronary disease at high risk for ventricular arrhythmia. N Engl J Med 1996; 335: 1933 –1940. 21 Bigger JT. Prophylactic use of implanted cardiac defibrillators in patients at high risk for ventricular arrhythmias after coronary-artery bypass graft surgery. N Engl J Med 1997; 337: 1569 –1575. 22 Moss AJ, Hall WJ, Cannom DS, Daubert JP et al. Improved survival with an implanted defibrillator in patients with coronary disease at high risk for ventricular arrhythmia. N Engl J Med 1996; 335: 1933 –1940. 23 Moss A, Zareba W, Hall WJ et al. Prophylactic implantation of a defibrillator in patients with myocardial infarction and reduced ejection fraction. N Engl J Med 2002; 346: 877–883. 24 Moss AJ. Implantable cardioverter defibrillator therapy: the sickest patients benefit the most. Circulation 2000; 101: 1638 –1640. 25 Moss AJ, Fadl Y, Zareba W, Cannom DS, Hall WJ. Survival benefit with an implanted defibrillator in relation to mortality risk in chronic coronary heart disease. Am J Cardiol 2001; 88: 516 –520. 26 Buxton AE, Lee KL, Fisher JD, Josephson ME, Prystowsky EU, Hafley G. A randomized study of the prevention of sudden death in patients with coronary artery disease. N Engl J Med 1999; 341: 1882 –1890. 27 Nisam S, Breithardt G. Lessons learned from neutral ICD trials. Circulation in press. 28 Bänsch D, Antz M, Boczor S, Volkmer M et al. Primary prevention of sudden cardiac death in idiopathic dilated cardiomyopathy: the Cardiomyopathy Trial (CAT). Circulation 2002; 105: 1453 –1458. TETC32 12/2/05 14:26 Page 969 Ventricular Tachycardia 29 Bänsch D, Böcker D, Brunn J, Weber M, Breithardt G, Block M. Clusters of ventricular tachycardias signify impaired survival in patients with idiopathic dilated cardiomyopathy and implantable cardioverter defibrillators. J Am Coll Cardiol 2000; 36: 566 –573. 30 Kadish A, Quigg R, Schaechter A, Anderson KP, Estes M, Levine J. Defibrillators in nonischemic cardiomyopathy treatment evaluation. Pacing Clin Electrophysiol 2000; 23: 338–343. 31 Kadish A, Dyer A, Daubert JP, Quigg R, Estes M. Prophylactic defibrillator implantation in patients with nonischemic dilated cardiomyopathy. N Engl J Med 2004; 350: 2151–2158. 32 Strickberger SA, Hummel JD, Bartlett TG et al. Amiodarone versus implantable cardioverter-defibrillator: randomized trial in patients with nonischemic dilated cardiomyopathy and asymptomatic nonsustained ventricular tachycardia—AMIOVIRT. J Am Coll Cardiol 2003; 41: 1707–1712. 33 Massie BM, Fisher SG, Deedwania PC, Singh BN, Fletcher RD, Singh SN. Effect of amiodarone on clinical status and left ventricular function in patients with congestive heart failure. Circulation 1996; 93: 2128 –2134. 34 Grimm W, Christ M, Bach J, Muller HH, Maisch B. Non-invasive arrhythmia risk stratification in idiopathic dilated cardiomyopathy: results of the Marburg Cardiomyopathy Study. Circulation 2003; 108: 2883–2891. 35 Borggrefe M, Podczeck A, Ostermeyer J, Breihardt G. Long-term results of electrophysiologically guided antitachycardia surgery in ventricular tachyarrhythmias: a collaborative report on 665 patients. In: Breihardt G, Borggrefe M, Zipes D, eds. Nonpharmacological Therapy of Tachyarrhythmias. Mount Kisco, New York: Futura Publishing; 1987: 109 –132. 36 Marcus FI, Fontaine GH, Guiraudon G et al. Right ventricular dysplasia: a report of 24 adult cases. Circulation 1982; 65: 384 –398. 37 Corrado D, Fontaine G, Marcus FI et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: need for an international registry. Study Group on Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy of the Working Groups on Myocardial and Pericardial Disease and Arrhythmias of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the World Heart Federation. Circulation 2000; 101: E101–E106. 38 Thiene G, Nava A, Corrado D, Rossi L, Pennelli N. Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med 1988; 318: 129 –133. 39 McKenna WJ, Thiene G, Nava A et al. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology. Br Heart J 1994; 71: 215 –218. 40 Corrado D, Leoni L, Link MS et al. Implantable cardioverter-defibrillator therapy for prevention of sudden death in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation 2003; 108: 3084 –3091. 41 Wichter T, Paul M, Wollmann C et al. Implantable cardioverter/defibrillator therapy in arrhythmogenic right ventricular cardiomyopathy: single-center experience of long-term follow-up and complications in 60 patients. Circulation 2004; 109: 1503 –1508. 42 Wigle ED, Rakowski H, Kimball BP, Williams WG. Hypertrophic cardiomyopathy. Clinical spectrum and treatment. Circulation 1995; 92: 1680 –1692. 43 Maron BJ, McKenna WJ, Danielson GK et al. American College of Cardiology/European Society of Cardiology clinical expert consensus document on hypertrophic cardiomyopathy. A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. J Am Coll Cardiol 2003; 42: 1687–1713. 44 Maron BJ, Shen WK, Link MS et al. Efficacy of implantable cardioverter-defibrillators for the prevention of sudden death in patients with hypertrophic cardiomyopathy. N Engl J Med 2000; 342: 365 –373. 45 McKenna WJ, Behr ER. Hypertrophic cardiomyopathy: management, risk stratification, and prevention of sudden death. Heart 2002; 87: 169 –176. 46 Maron BJ, Estes NA, III, Maron MS, Almquist AK, Link MS, Udelson JE. Primary prevention of sudden death as a novel treatment strategy in hypertrophic cardiomyopathy. Circulation 2003; 107: 2872 –2875. 47 Gallavardin L. Extrasystolie ventriculaire a paroxysmes tachycardiques prolonges. Arch Mal Coeur Vaiss 1922; 15: 298–306. 48 Moss AJ, Schwartz PJ, Crampton RS, Locati E, Carleen E. The long QT syndrome: a prospective international study. Circulation 1985; 71: 17–21. 49 Moss AJ, Schwartz PJ, Crampton RS et al. The long QT syndrome. Prospective longitudinal study of 328 families. Circulation 1991; 84: 1136 –1144. 50 Schwartz PJ, Locati E. The idiopathic long QT syndrome: pathogenetic mechanisms and therapy. Eur Heart J 1985; 6: 103 –114. 51 Camm AJ, Janse MJ, Roden DM, Rosen MR, Cinca J, Cobbe SM. Congenital and acquired long QT syndrome. Eur Heart J 2000; 21: 1232 –1237. 52 Haverkamp W, Breithardt G, Camm AJ et al. The potential for QT prolongation and proarrhythmia b y non-antiarrhythmic drugs: clinical and regulatory implications. Report on a policy conference of the European Society of Cardiology. Eur Heart J 2000; 21: 1216 –1231. 53 Eckardt L, Haverkamp W, Borggrefe M, Breithardt G. Experimental models of torsade de pointes. Cardiovasc Res 1998; 39: 178 –193. 54 Moss AJ, Zareba W, Benhorin J et al. ECG T-wave patterns 969 TETC32 12/2/05 14:26 Page 970 970 Chapter 32 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 in genetically distinct forms of the hereditary long QT syndrome. Circulation 1995; 92: 2929–2934. Priori SG, Schwartz PJ, Napolitano C et al. Risk stratification in the long-QT syndrome. N Engl J Med 2003; 348: 1866 –1874. Schwartz PJ, Priori SG, Spazzolini C et al. Genotypephenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation 2001; 103: 89 –95. Zareba W, Moss AJ, Locati EH et al. Modulating effects of age and gender on the clinical course of long QT syndrome by genotype. J Am Coll Cardiol 2003; 42: 103–109. Priori SG, Aliot E, Blomstrom-Lundqvist C et al. Task Force on Sudden Cardiac Death, European Society of Cardiology. Europace 2002; 4: 3–18. Chiang CE, Roden DM. The long QT syndromes: genetic basis and clinical implications. J Am Coll Cardiol 2000; 36: 1–12. Moss AJ, Zareba W, Hall WJ et al. Effectiveness and limitations of beta-blocker therapy in congenital long-QT syndrome. Circulation 2000; 101: 616 – 623. Sato T, Hata Y, Yamamoto M et al. Early afterdepolarization abolished by potassium channel opener in a patient with idiopathic long QT syndrome. J Cardiovasc Electrophysiol 1995; 6: 279 –282. Shimizu W, Kurita T, Matsuo K et al. Improvement of repolarization abnormalities by a K+ channel opener in the LQT1 form of congenital long-QT syndrome. Circulation 1998; 97: 1581–1588. Wang DW, Yazawa K, Makita N, George-AL J, Bennett PB. Pharmacological targeting of long QT mutant sodium channels. J Clin Invest 1997; 99: 1714 –1720. Kehl HG, Haverkamp W, Rellensmann G et al. Images in cardiovascular medicine. Life-threatening neonatal arrhythmia: successful treatment and confirmation of clinically suspected extreme long QT-syndrome-3. Circulation 2004; 109: e205 –e206. An RH, Bangalore R, Rosero SZ, Kass RS. Lidocaine block of LQT-3 mutant human Na+ channels. Circ Res 1996; 79: 103–108. Priori SG, Napolitano C, Cantu F, Brown AM, Schwartz PJ. Differential response to Na+ channel blockade, betaadrenergic stimulation, and rapid pacing in a cellular model mimicking the SCN5A and HERG defects present in the long-QT syndrome. Circ Res 1996; 78: 1009 –1015. Benhorin J, Taub R, Goldmit M et al. Effects of flecainide in patients with new SCN5A mutation: mutation-specific therapy for long-QT syndrome? Circulation 2000; 101: 1698 –1706. Zareba W, Moss AJ, Daubert JP, Hall WJ, Robinson JL, Andrews M. Implantable cardioverter defibrillator in high-risk long QT syndrome patients. J Cardiovasc Electrophysiol 2003; 14: 337–341. Viskin S. Implantable cardioverter defibrillator in highrisk long QT syndrome patients. J Cardiovasc Electrophysiol 2003; 14: 1130 –1131. Gussak I, Brugada P, Brugada J et al. Idiopathic short QT interval: a new clinical syndrome? Cardiology 2000; 94: 99–102. 71 Gaita F, Giustetto C, Bianchi F et al. Short QT Syndrome: a familial cause of sudden death. Circulation 2003; 108: 965–970. 72 Brugada R, Hong K, Dumaine R et al. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation 2004; 109: 30 –35. 73 Schimpf R, Wolpert C, Bianchi F et al. Congenital short QT syndrome and implantable cardioverter defibrillator treatment: inherent risk for inappropriate shock delivery. J Cardiovasc Electrophysiol 2003; 14: 1273–1277. 74 Viskin S, Belhassen B. Polymorphic ventricular tachyarrhythmias in the absence of organic heart disease: classification, differential diagnosis, and implications for therapy. Prog Cardiovasc Dis 1998; 41: 17–34. 75 Swan H, Laitinen PJ. Familial polymorphic ventricular tachycardia: intracellular calcium channel disorder. Card Electrophysiol Rev 2002; 6: 81–87. 76 Priori SG, Napolitano C, Memmi M et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation 2002; 106: 69 –74. 77 Marks AR, Priori S, Memmi M, Kontula K, Laitinen PJ. Involvement of the cardiac ryanodine receptor/calcium release channel in catecholaminergic polymorphic ventricular tachycardia. J Cell Physiol 2002; 190: 1–6. 78 Laitinen PJ, Brown KM, Piippo K et al. Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation 2001; 103: 485 –490. 79 Bauce B, Rampazzo A, Basso C et al. Screening for ryanodine receptor type 2 mutations in families with effort-induced polymorphic ventricular arrhythmias and sudden death: early diagnosis of asymptomatic carriers. J Am Coll Cardiol 2002; 40: 341–349. 80 Lahat H, Eldar M, Levy-Nissenbaum E et al. Autosomal recessive catecholamine- or exercise-induced polymorphic ventricular tachycardia: clinical features and assignment of the disease gene to chromosome 1p13–21. Circulation 2001; 103: 2822 –2827. 81 Postma AV, Denjoy I, Hoorntje TM et al. Absence of calsequestrin 2 causes severe forms of catecholaminergic polymorphic ventricular tachycardia. Circ Res 2002; 91: e21–e26. 82 Priori SG, Napolitano C, Tiso N et al. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation 2001; 103: 196 –200. 83 Fisher JD, Krikler D, Hallidie-Smith KA. Familial polymorphic ventricular arrhythmias: a quarter century of successful medical treatment based on serial exercisepharmacologic testing. J Am Coll Cardiol 1999; 34: 2015 –2022. 84 Lahat H, Pras E, Olender T et al. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am J Hum Genet 2001; 69: 1378 –1384. 85 Survivors of out-of-hospital cardiac arrest with apparently normal heart. Need for definition and standardized TETC32 12/2/05 14:26 Page 971 Ventricular Tachycardia 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 clinical evaluation. Consensus Statement of the Joint Steering Committees of the Unexplained Cardiac Arrest Registry of Europe and of the Idiopathic Ventricular Fibrillation Registry of the United States. Circulation 1997; 95: 265–272. Belhassen B, Viskin S. Idiopathic ventricular tachycardia and fibrillation. J Cardiovasc Electrophysiol 1993; 4: 356–368. Priori SG, Napolitano C, Grillo M. Concealed arrhythmogenic syndromes: the hidden substrate of idiopathic ventricular fibrillation? Cardiovasc Res 2001; 50: 218–223. Viskin S, Belhassen B. Idiopathic ventricular fibrillation. Am Heart J 1990; 120: 661– 671. Haissaguerre M, Shoda M, Jais P et al. Mapping and ablation of idiopathic ventricular fibrillation. Circulation 2002; 106: 962 – 967. Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol 1992; 20: 1391–1396. Nademanee K, Veerakul G, Nimmannit S et al. Arrhythmogenic marker for the sudden unexplained death syndrome in Thai men. Circulation 1997; 96: 2595–2600. Wilde AA, Antzelevitch C, Borggrefe M et al. Proposed diagnostic criteria for the Brugada syndrome: consensus report. Circulation 2002; 106: 2514 –2519. Chen Q, Kirsch GE, Zhang D et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 1998; 392: 293 –296. Schulze-Bahr E, Eckardt L, Breithardt G et al. Sodium channel gene (SCN5A) mutations in 44 index patients with Brugada syndrome: different incidences in familial and sporadic disease. Hum Mutat 2003; 21: 651–652. Smits JP, Eckardt L, Probst V et al. Genotype-phenotype relationship in Brugada syndrome: electrocardiographic features differentiate SCN5A-related patients from nonSCN5A-related patients. J Am Coll Cardiol 2002; 40: 350–356. Brugada R, Brugada J, Antzelevitch C et al. Sodium channel blockers identify risk for sudden death in patients with ST-segment elevation and right bundle branch block but structurally normal hearts. Circulation 2000; 101: 510–515. Rolf S, Bruns HJ, Wichter T et al. The ajmaline challenge in Brugada syndrome: diagnostic impact, safety, and recommended protocol. Eur Heart J 2003; 24: 1104 –1112. Miyazaki T, Mitamura H, Miyoshi S et al. Autonomic and antiarrhythmic drug modulation of ST segment elevation in patients with Brugada syndrome. J Am Coll Cardiol 1996; 27: 1061–1070. Antzelevitch C, Brugada R. Fever and Brugada syndrome. Pacing Clin Electrophysiol 2002; 25: 1537–1539. Brugada J, Brugada R, Brugada P. Determinants of sudden cardiac death in individuals with the electrocardiographic pattern of Brugada syndrome and no previous cardiac arrest. Circulation 2003; 108: 3092 –3096. Eckardt L, Kirchhof P, Schulze-Bahr E et al. 102 103 104 105 106 107 108 109 110 111 112 113 114 115 Electrophysiologic investigation in Brugada syndrome: yield of programmed ventricular stimulation at two ventricular sites with up to three premature beats. Eur Heart J 2002; 23: 1394 –1401. Priori SG, Napolitano C, Gasparini M et al. Natural history of Brugada syndrome: insights for risk stratification and management. Circulation 2002; 105: 1342–1347. Eckardt L, Probst V, Smits JP, Schulze-Bahr E et al. Longterm prognosis of individuals with right precordial STelevation—Brugada syndrome. Circulation; in press. Brugada J, Brugada R, Brugada P. Right bundle-branch block and ST-segment elevation in leads V1 through V3: a marker for sudden death in patients without demonstrable structural heart disease. Circulation 1998; 97: 457–460. Belhassen B, Viskin S, Antzelevitch C. The Brugada syndrome: is an implantable cardioverter defibrillator the only therapeutic option? Pacing Clin Electrophysiol 2002; 25: 1634 –1640. Belhassen B, Glick A, Viskin S. Efficacy of quinidine in high-risk patients with Brugada syndrome. Circulation 2004; 110: 1731–1737. Belhassen B, Viskin S, Fish R et al. Effects of electrophysiologic-guided therapy with Class IA antiarrhythmic drugs on the long-term outcome of patients with idiopathic ventricular fibrillation with or without the Brugada syndrome. J Cardiovasc Electrophysiol 1999; 10: 1301–1312. Brugada J, Brugada R, Antzelevitch C et al. Long-term follow-up of individuals with the electrocardiographic pattern of right bundle-branch block and ST-segment elevation in precordial leads V1 to V3. Circulation 2002; 105: 73–78. O’Donnell D, Cox D, Bourke J et al. Clinical and electrophysiological differences between patients with arrhythmogenic right ventricular dysplasia and right ventricular outflow tract tachycardia. Eur Heart J 2003; 24: 801–810. Lerman BB, Stein K, Engelstein ED et al. Mechanism of repetitive monomorphic ventricular tachycardia. Circulation 1995; 92: 421–429. Mont L, Seixas T, Brugada P et al. Clinical and electrophysiologic characteristics of exercise-related idiopathic ventricular tachycardia. Am J Cardiol 1991; 68: 897–900. Mont L, Seixas T, Brugada P et al. The electrocardiographic, clinical, and electrophysiologic spectrum of idiopathic monomorphic ventricular tachycardia. Am Heart J 1992; 124: 746 –753. Gill JS, Blaszyk K, Ward DE, Camm AJ. Verapamil for the suppression of idiopathic ventricular tachycardia of left bundle branch block-like morphology. Am Heart J 1993; 126: 1126 –1133. Ribbing M, Wasmer K, Monnig G et al. Endocardial mapping of right ventricular outflow tract tachycardia using noncontact activation mapping. J Cardiovasc Electrophysiol 2003; 14: 602 –608. Ohe T, Shimomura K, Aihara N et al. Idiopathic sustained left ventricular tachycardia: clinical and 971 TETC32 12/2/05 14:26 Page 972 972 Chapter 32 116 117 118 119 120 electrophysiologic characteristics. Circulation 1988; 77: 560–568. Lin FC, Finley CD, Rahimtoola SH, Wu D. Idiopathic paroxysmal ventricular tachycardia with a QRS pattern of right bundle branch block and left axis deviation: a unique clinical entity with specific properties. Am J Cardiol 1983; 52: 95 –100. Nakagawa H, Beckman KJ, McClelland JH et al. Radiofrequency catheter ablation of idiopathic left ventricular tachycardia guided by a Purkinje potential. Circulation 1993; 88: 2607–2617. Rosenbaum MB, Blanco HH, Elizari MV et al. Electrotonic modulation of the T wave and cardiac memory. Am J Cardiol 1982; 50: 213 –222. Blanchot P, Warin JF. [A further case of ventricular tachycardia by re-entry]. Arch Mal Coeur Vaiss 1973; 66: 915–923. Touboul P, Claveyrolas R, Huerta F, Porte J, Delahaye JP. [Ventricular tachycardia due to premature supra- 121 122 123 124 ventricular beats with a normal QRS complex. Analysis of a case]. Arch Mal Coeur Vaiss 1975; 68: 969 –976. Ward DE, Nathan AW, Camm AJ. Fascicular tachycardia sensitive to calcium antagonists. Eur Heart J 1984; 5: 896–905. Kottkamp H, Chen X, Hindricks G et al. Idiopathic left ventricular tachycardia: new insights into electrophysiological characteristics and radiofrequency catheter ablation. Pacing Clin Electrophysiol 1995; 18: 1285 –1297. German LD, Packer DL, Bardy GH, Gallagher JJ. Ventricular tachycardia induced by atrial stimulation in patients without symptomatic cardiac disease. Am J Cardiol 1983; 52: 1202 –1207. Eckardt L, Breithardt G, Haverkamp W. Idiopathic left ventricular tachycardia localized at the distal end of the posterior fascicle by non-contact activation mapping. Heart 2002; 87: 374.