Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Management of acute coronary syndrome wikipedia , lookup
Remote ischemic conditioning wikipedia , lookup
Coronary artery disease wikipedia , lookup
Hypertrophic cardiomyopathy wikipedia , lookup
Electrocardiography wikipedia , lookup
Rheumatic fever wikipedia , lookup
Cardiac contractility modulation wikipedia , lookup
Quantium Medical Cardiac Output wikipedia , lookup
Cardiac surgery wikipedia , lookup
Dextro-Transposition of the great arteries wikipedia , lookup
Heart failure wikipedia , lookup
Arrhythmogenic right ventricular dysplasia wikipedia , lookup
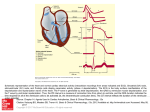
See related article, pages 638 – 645 A Surprising New Arrhythmia Mechanism in Heart Failure Dan M. Roden H Downloaded from http://circres.ahajournals.org/ by guest on October 14, 2016 eart failure is the most common discharge diagnosis in the United States, with a prognosis worse than many cancers.1 Patients with heart failure die for two reasons: advanced circulatory insufficiency or sudden death.2 We know that continuous online monitoring of cardiac rhythm from patients with advanced heart failure is inevitably abnormal and often shows long runs of irregular and/or polymorphic ventricular tachycardia. Unlike patients with ventricular tachycardia due to healed myocardial infarction, those with heart failure rarely have ventricular tachycardia inducible by programmed stimulation, and when it is, its prognostic importance is uncertain. Therapy with conventional antiarrhythmics is no more successful in patients with heart failure than in those with serious ventricular arrhythmias due to healed infarction. notypes in cellular, animal, and human models include calcium-mediated activation of transcriptional programs,7,8 a leaky calcium release channel,9,10 and increased CaM kinase activity.11 Another common electrophysiological finding both in animal models and in patients with heart failure is reduction in a range of potassium currents, including ITO, IK1, and the delayed rectifier components.12 Indeed, this effect, which in at least some cases has been definitively attributed to decreased ion channel gene transcription,13 contributes to action potential prolongation and disordered QT regulation, another characteristic finding in heart failure.14,15 Marbán, Tomaselli, and colleagues proposed in the mid-1990s16 that such action potential prolongation would be linked to arrhythmogenesis, in effect that a torsades de pointes–like mechanism (at the time not well defined) might be linked to the problem of serious arrhythmias in heart failure. In this issue of Circulation Research, Akar and Rosenbaum provide evidence for that link,17 by reporting striking similarities in the electrophysiological properties of “wedge preparations” isolated from dogs with pacing-induced heart failure, compared with those from animals treated with drugs causing torsades de pointes. Arrhythmias, Disordered [Ca2ⴙ]i Homeostasis, and Kⴙ Current Downregulation in Heart Failure Given the huge public health impact of the problem and the failure of current pharmacological therapies, one obvious way forward in attacking the problem of sudden death in heart failure is to understand the basic molecular mechanisms underlying arrhythmia susceptibility in this setting. The disordered contractility that characterizes heart failure naturally suggests abnormal intracellular calcium homeostasis, a well-recognized feature of the heart failure phenotype,3,4 as a candidate arrhythmia mechanism, and experimental studies support this idea. Pogwizd and colleagues have used threedimensional activation mapping in rabbits with volumeoverload heart failure to identify focal triggers and repetitive focal activity as a major mechanism underlying VT in this model, 5 behaviors that are highly reminiscent of afterdepolarization-related triggering due to intracellular calcium overload. Indeed, their further studies indicated that heart failure–related upregulation of the sodium-calcium exchanger, in the setting of downregulation of the inward rectifier and maintained -adrenergic responsiveness, markedly increases the propensity for triggered arrhythmias due to intracellular calcium overload.6 More upstream mechanisms implicated as drivers of the “hypertrophy/heart failure” phe- The Wedge Preparation and Transmural Heterogeneity of Repolarization Seminal observations from the laboratory of Antzelevitch over the past decade have documented the existence of a previously unrecognized cell type in the midmyocardium whose unusual response to electrophysiological stressors such as proarrhythmic drugs is an important contributor to long QT–associated arrhythmias.18 –20 Indeed, M cells may constitute up to 40% of the bulk of the ventricular myocardium. The left ventricular “wedge” preparation, originated and popularized by the Antzelevitch laboratory, has identified marked drug-induced action potential prolongation in the M-cell layer as a key contributor to increased transmural heterogeneity of repolarization that, in turn, appears to generate the substrate for reentry that is now thought to underlie typical torsades de pointes.21 The concept that heterogeneity, or “dispersion,” of action potential durations creates a substrate for reentry is an older one, but the concept that this heterogeneity might extend to sites across the ventricular wall, rather than between adjacent sites located in the same cell layer, is newer. In the heart failure wedge preparation, as in the drugtreated wedge preparation, there was marked prolongation of action potential durations among M cells, with much less prolongation in endocardial and epicardial cell types, leading to increased dispersion of repolarization. Further, in both settings, stimulation from the epicardium (the site with the shortest action potentials) generates conduction block at the The opinions expressed in this editorial are not necessarily those of the editors or of the American Heart Association. From the Division of Clinical Pharmacology, Vanderbilt University School of Medicine, Nashville, Tenn. Correspondence to Dan M. Roden, MD, Professor of Medicine and Pharmacology, Director, Division of Clinical Pharmacology, Vanderbilt University School of Medicine, 532 Medical Research Building I, Nashville, TN 37232. E-mail [email protected] (Circ Res. 2003;93:589-591.) © 2003 American Heart Association, Inc. Circulation Research is available at http://www.circresaha.org DOI: 10.1161/01.RES.0000095382.50153.7D 589 590 Circulation Research October 3, 2003 Downloaded from http://circres.ahajournals.org/ by guest on October 14, 2016 epicardial-midmyocardial junction with polymorphic arrhythmias arising as a result of conduction around this region of functional block. Extending the link to heart failure are studies in the wedge and other preparations that have implicated disordered intracellular calcium homeostasis as a potential contributor to drug-induced torsades de pointes.22,23 M cells display long basal action potentials, suggesting reduced net inward current, and this may underlie their susceptibility to display such marked prolongation with QT-prolonging drugs (or pacing-induced heart failure). However, it is not yet established why M cells display long action potentials under normal conditions; suggested possibilities include reduced K⫹ current, increased inward current, altered intracellular calcium handling, or altered gap junction protein function.17,24,25 Most importantly, we have yet to understand how a disease like heart failure, however defined, engenders changes in expression or function of not only these candidate proteins, but a myriad of others that together make up the complex signaling pathways that we call normal electrophysiology. Even more fundamentally, we do not understand the signals that tell a cell to become an M cell or the mechanisms that determine their unusual, and not totally homogeneous, distribution within the midmyocardium; obviously, their location and their electrophysiological properties will be linked in some as yet poorly understood fashion. Indeed, a difficulty with this field is an exact definition of what constitutes a normal M cell. It is a cell with a long action potential, like those in the endocardium, but with a prominent phase 1 notch, as in the epicardium. These gross morphological descriptors of action potential configuration reflect balancing acts among a multitude of inward and outward currents, but how big a notch constitutes a notch or how long an action potential one must have before an M cell becomes an M cell and is no longer an epicardial cell is not so clear. The gradients described by Akar and Rosenbaum17 suggest that these distinctions are continuous rather than abrupt, and thus we should expect gradients of expression of channels and other proteins important for electrogenesis in the same fashion. Implications for Heart Failure Research and Patient Care Any investigator studying heart failure or any clinician caring for patients with heart failure will immediately recognize that the diagnosis of “heart failure” itself represents a highly heterogeneous mixture of clinical diagnoses, presentations, and responses to therapies. Thus, an interesting and important question raised by the present study is how commonly increased transmural dispersion of repolarization occurs in heart failure and to what extent it contributes to generating the associated arrhythmia-prone substrate. As a corollary, one can turn the question around: having implicated heterogeneity of repolarization as an important mechanism in clinically important polymorphic tachycardias such as classical torsades de pointes and ventricular tachycardia in heart failure, how often does this mechanism underlie other polymorphic tachycardias? Many insults cause the contractile dysfunction/arrhythmia phenotype we call heart failure. Intervening once this pheno- type is in place has the appeal that the disease is readily recognized, and many therapies aimed at symptoms—regardless of the underlying cause—provide some benefit. However, early intervention to correct the molecular dysfunction that leads to arrhythmias in this setting seems more appealing. This presents the problem that we have to think about advanced clinical phenotypes like heart failure as many separate diseases, each with its own molecular triggers and specific therapies that should be curative if applied early. This is not so different from the way oncologists are approaching the problem of “cancer.” Finally, what does any of this mean for patient care? One interesting characteristic of the wedge preparation is that polymorphic ventricular tachycardia is most readily induced by programmed electrical stimulation from the site with the shortest action potentials, the epicardium. Whether this is the case in patients is not certain. In whole animal models of torsades de pointes, the arrhythmias have been reported to be initiated by endocardial foci, possibly representing afterdepolarizations in the Purkinje network.26 More recently, a proarrhythmic effect of epicardial pacing (as now increasingly used clinically in patients with heart failure and biventricular pacing devices) has been reported.27 The early experience with biventricular pacing, however, suggests that overall mortality is not increased.28 Is it possible that techniques of programmed stimulation using epicardial approaches could be used to stratify arrhythmia risk among patients with heart failure? Or patients with the long-QT syndromes, where conventional electrophysiological testing (that uses endocardial stimulation) has also not proven terribly helpful? Thus, description of an unexpected role of exaggerated transmural heterogeneity of repolarization in this study identifies both clinical opportunities as well as an interesting new starting point for development and evaluation of new therapies to deal with the challenge of sudden death in heart failure. Acknowledgments This work was supported in part by grants from the United States Public Health Service (HL46681, HL49989, and HL65962). Dr Roden is the holder of the William Stokes Chair in Experimental Therapeutics, a gift from the Dai-ichi Corporation. References 1. Stewart S, MacIntyre K, Hole DJ, Capewell S, McMurray JJ. More “malignant” than cancer? Five-year survival following a first admission for heart failure. Eur J Heart Fail. 2001;3:315–322. 2. Stevenson WG, Stevenson LW, Middlekauff HR, Fonarow GC, Hamilton MA, Woo MA, Saxon LA, Natterson PD, Steimle A, Walden JA. Improving survival for patients with advanced heart failure: a study of 737 consecutive patients. J Am Coll Cardiol. 1995;26:1417–1423. 3. Gwathmey JK, Copelas L, MacKinnon R, Schoen FJ, Feldman MD, Grossman W, Morgan JP. Abnormal intracellular calcium handling in myocardium from patients with end-stage heart failure. Circ Res. 1987; 61:70 –76. 4. Beuckelmann DJ, Näbauer M, Erdmann E. Intracellular calcium handling in isolated ventricular myocytes from patients with terminal heart failure. Circulation. 1992;85:1046 –1055. 5. Pogwizd SM, Corr PB. Reentrant and nonreentrant mechanisms contribute to arrhythmogenesis during early myocardial ischemia: results using three-dimensional mapping. Circ Res. 1987;61:352–371. 6. Pogwizd SM, Schlotthauer K, Li L, Yuan W, Bers DM. Arrhythmogenesis and contractile dysfunction in heart failure: roles of sodiumcalcium exchange, inward rectifier potassium current, and residual -adrenergic responsiveness. Circ Res. 2001;88:1159 –1167. Roden Downloaded from http://circres.ahajournals.org/ by guest on October 14, 2016 7. Sussman MA, Lim HW, Gude N, Taigen T, Olson EN, Robbins J, Colbert MC, Gualberto A, Wieczorek DF, Molkentin JD. Prevention of cardiac hypertrophy in mice by calcineurin inhibition. Science. 1998;281: 1690 –1693. 8. Nicol RL, Frey N, Olson EN. From the sarcomere to the nucleus: role of genetics and signaling in structural heart disease. Annu Rev Genomics Hum Genet. 2000;1:179 –223. 9. Marks AR. A guide for the perplexed: towards an understanding of the molecular basis of heart failure. Circulation. 2003;107:1456 –1459. 10. Wehrens XH, Lehnart SE, Huang F, Vest JA, Reiken SR, Mohler PJ, Sun J, Guatimosim S, Song LS, Rosemblit N, D’Armiento JM, Napolitano C, Memmi M, Priori SG, Lederer WJ, Marks AR. FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell. 2003;113:829 – 840. 11. Wu Y, Temple J, Zhang R, Dzhura I, Zhang W, Trimble R, Roden DM, Passier R, Olson EN, Colbran RJ, Anderson ME. Calmodulin kinase II and arrhythmias in a mouse model of cardiac hypertrophy. Circulation. 2002;106:1288 –1293. 12. Beuckelmann DJ, Näbauer M, Erdmann E. Alterations of K⫹ currents in isolated human ventricular myocytes from patients with terminal heart failure. Circ Res. 1993;73:379 –385. 13. Kaab S, Dixon J, Duc J, Ashen D, Nabauer M, Beuckelmann DJ, Steinbeck G, McKinnon D, Tomaselli GF. Molecular basis of transient outward potassium current downregulation in human heart failure: a decrease in Kv4.3 mRNA correlates with a reduction in current density. Circulation. 1998;98:1383–1393. 14. Berger RD, Kasper EK, Baughman KL, Marban E, Calkins H, Tomaselli GF. Beat-to-beat QT interval variability: novel evidence for repolarization lability in ischemic and nonischemic dilated cardiomyopathy. Circulation. 1997;96:1557–1565. 15. Barr CJ, Naas A, Freeman M, Lang CC, Struthers AD. QT dispersion and sudden unexpected death in chronic heart failure. Lancet. 1994;343: 327–329. 16. Tomaselli GF, Beuckelmann DJ, Calkins HG, Berger RD, Kessler PD, Lawrence JH, Kass D, Feldman AM, Marban E. Sudden cardiac death in heart failure: the role of abnormal repolarization. Circulation. 1994;90: 2534 –2539. 17. Akar FG, Rosenbaum DS. Transmural electrophysiological heterogeneities underlying arrhythmogenesis in heart failure. Circ Res. 2003;93: 638 – 645. 18. Sicouri S, Antzelevitch C. A subpopulation of cells with unique electrophysiological properties in the deep subepicardium of the canine ventricle: the M cell. Circ Res. 1991;68:1729 –1741. Arrhythmia Mechanisms in CHF 591 19. Shimizu W, Antzelevitch C. Differential effects of -adrenergic agonists and antagonists in LQT1, LQT2 and LQT3 models of the long QT syndrome. J Am Coll Cardiol. 2000;35:778 –786. 20. Akar FG, Yan GX, Antzelevitch C, Rosenbaum DS. Unique topographical distribution of M cells underlies reentrant mechanism of torsade de pointes in the long-QT syndrome. Circulation. 2002;105:1247–1253. 21. Yan GX, Shimizu W, Antzelevitch C. Characteristics and distribution of M cells in arterially perfused canine left ventricular wedge preparations. Circulation. 1998;98:1921–1927. 22. Burashnikov A, Antzelevitch C. Block of IKs does not induce early afterdepolarization activity but promotes -adrenergic agonist-induced delayed afterdepolarization activity. J Cardiovasc Electrophysiol. 2000; 11:458 – 465. 23. Wu Y, Roden DM, Anderson ME. Calmodulin kinase inhibition prevents development of the arrhythmogenic transient inward current. Circ Res. 1999;84:906 –912. 24. Zygmunt AC, Eddlestone GT, Thomas GP, Nesterenko VV, Antzelevitch C. Larger late sodium conductance in M cells contributes to electrical heterogeneity in canine ventricle. Am J Physiol Heart Circ Physiol. 2001;281:H689 –H697. 25. Liu DW, Antzelevitch C. Characteristics of the delayed rectifier current (IKr and IKs) in canine ventricular epicardial, midmyocardial, and endocardial myocytes: a weaker IKs contributes to the longer action potential of the M cell. Circ Res. 1995;76:351–365. 26. El-Sherif N, Caref EB, Yin H, Restivo M. The electrophysiological mechanism of ventricular arrhythmias in the long QT syndrome: tridimensional mapping of activation and recovery patterns. Circ Res. 1996; 79:474 – 492. 27. Medina-Ravell VA, Lankipalli RS, Yan GX, Antzelevitch C, MedinaMalpica NA, Medina-Malpica OA, Droogan C, Kowey PR. Effect of epicardial or biventricular pacing to prolong QT interval and increase transmural dispersion of repolarization: does resynchronization therapy pose a risk for patients predisposed to long QT or torsade de pointes? Circulation. 2003;107:740 –746. 28. Young JB, Abraham WT, Smith AL, Leon AR, Lieberman R, Wilkoff B, Canby RC, Schroeder JS, Liem LB, Hall S, Wheelan K. Combined cardiac resynchronization and implantable cardioversion defibrillation in advanced chronic heart failure: the MIRACLE ICD trial. JAMA. 2003; 289:2685–2694. KEY WORDS: arrhythmia 䡲 heart failure 䡲 long QT A Surprising New Arrhythmia Mechanism in Heart Failure Dan M. Roden Downloaded from http://circres.ahajournals.org/ by guest on October 14, 2016 Circ Res. 2003;93:589-591 doi: 10.1161/01.RES.0000095382.50153.7D Circulation Research is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231 Copyright © 2003 American Heart Association, Inc. All rights reserved. Print ISSN: 0009-7330. Online ISSN: 1524-4571 The online version of this article, along with updated information and services, is located on the World Wide Web at: http://circres.ahajournals.org/content/93/7/589 Permissions: Requests for permissions to reproduce figures, tables, or portions of articles originally published in Circulation Research can be obtained via RightsLink, a service of the Copyright Clearance Center, not the Editorial Office. Once the online version of the published article for which permission is being requested is located, click Request Permissions in the middle column of the Web page under Services. Further information about this process is available in the Permissions and Rights Question and Answer document. Reprints: Information about reprints can be found online at: http://www.lww.com/reprints Subscriptions: Information about subscribing to Circulation Research is online at: http://circres.ahajournals.org//subscriptions/