Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
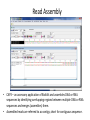
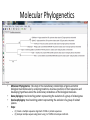
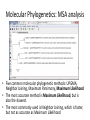
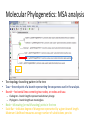
Introduction to Bioinformatics Resources for DNA Barcoding DNA Barcoding • • • • DNA barcoding is a tool for rapid species identification based on DNA sequences DNA barcodes consist of a standardized short sequence of DNA (400–800 bp) that in principle should be easily generated and characterized for all species on the planet. DNA barcoding aims to use the information of one or a few gene regions to identify all species of life, General Steps: – – – – DNA amplification of DNA fragment DNA sequencing and assembly species identification molecular phylogenetic analysis http://jeremydewaard.com/wpcontent/uploads/2010/01/Floyd_et_al_fig_1.png Bioinformatics http://medvetande.dk/images/CentralDogmBiomole cule_2000.jpg http://labs.gladstone.ucsf.edu/bioinformatics/sites/default/files/i magecache/os_modal_image_300/bioinformatics/files/bioinfor.j pg Bioinformatics is defined as an interdisciplinary research area that applies computer and information science to solve biological problems. DNA Sequencing http://www.nsf.gov/news/mmg/media/images/maize_sequence_f.jpg DNA Sequence Trace • Four color chromatogram showing the results of a sequencing run. • The characters below each peak represents the softwares attempt at identifying the correct nucleotide. • Errors commonly occur near the beginning and the end of any read. Read Assembly • CAP3 – an accessory application of BioEdit and assembles DNA or RNA sequences by identifying overlapping regions between multiple DNA or RNA sequences and merges (assembles) them. • Assembled reads are referred to as contigs, short for contiguous sequence. Basic Local Alignment Search Tool (BLAST) • Compares a query sequence to a database collection of sequences. • Retrieves significantly similar sequences • Blast tools: blastn, blastp, blastx, tblastn, tblastx Multiple Sequence Alignments • An alignment between 3 or more sequences • The algorithm identifies a series of characters that are in the same order in both sequences. • The assumption is that all sequences in a multiple sequence alignment are evolutionarily related. • Highlights insertion/deletion and amino acid substitution events Molecular Phylogenetics • • • • Molecular Phylogenetics: the study of the evolutionary relationships of genes and other biological macromolecules by analyzing mutations at various positions in their sequences and developing hypotheses about the evolutionary relatedness of the biological molecules. Gene phylogeny: tree branching pattern representing the evolution of a group of related genes Species phylogeny: tree branching pattern representing the evolution of a group of related species Steps: – – (1) Create a multiple sequence alignment of DNA or protein sequences (2) Analyze multiple sequence alignment using 1 of 5 different analyses methods. Molecular Phylogenetics: MSA analysis • Five common molecular phylogenetic methods: UPGMA, Neighbor Joining, Maximum Parsimony, Maximum Likelihood • The most accurate method is Maximum Likelihood, but is also the slowest. • The most commonly used is Neighbor Joining, which is faster, but not as accurate as Maximum Likelihood Molecular Phylogenetics: MSA analysis • • • Tree topology: branching pattern in the tree Taxa – the end point of a branch representing the sequences used in the analysis. Branch – horizontal lines conneting two nodes, or nodes and taxa – Cladogram – branch lengths represent evolutionary change – Phylogram – branch lengths are meaningless • • Node – bifurcating (or multifurcating) points in the tree Scale Bar – indicates degree of divergence represented by a given branch length. Maximum Likelihood measures average number of substitutions per site. Molecular Phylogenetics: MSA analysis • Bootstrapping: statistical technique that tests the sampling errors of a phylogenetic tree. • Bootstrap values are measures of confidence of the tree topology. The higher the value the more the relationship can be trusted. The End • Lets do a little bioinformatics