Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
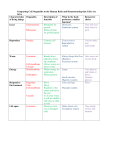
MCB Lecture 6 – Lysosomes What is a Primary Lysosome? o They come directly off of the Trans-Golgi and contain newly synthesized enzymes. They have not yet acquired material to be digested. What is a Secondary Lysosome? o Once a Primary Lysosome fuses with substrate to degrade, it becomes a Secondary Lysosome. What is the difference between an Early Endosome and a Late Endosome? o Endosomes form when vesicles from the Plasma Membrane come into the cell and fuse. An Early Lysosome has a lot of cytoplasm, little enzymes, and is less concentrated with material. A Late Endosome has lost cytoplasm and membrane (it is recycled) and it fuses with lysosomes and becomes more and more acidic to degrade the contents inside. What happens when an Endosome “matures”? o Increases acidity (decreases pH) and increases concentration (decreases in size). Under what circumstances do vesicles get a Clathrin Coat? o Vesicles budding off of the Golgi to go to a Lysosome of an Endosome. o Vesicles coming inside of the cell from outside of the cell and fusing with Lysosomes or Endosomes What are proteins tagged with that go to the Lysosome? Where do they get this tag? o Mannose-6-Phosphate o Tagged in the Cis-Golgi What is the enzyme that adds on the M-6-P Tag? o N-Acetylglucosamine Phosphotransferase Coated Vesicles: o When a protein goes from the ER to the Golgi, what protein is used to coat? COP II o When a protein goes from the Golgi to the ER, what protein is used to coat? COP I o When endocytosis is occurring, what protein is used to coat? Clathrin o When proteins go from the Golgi to the Lysosome and back, what protein is used to coat? Clathrin What are Hydrolytic enzymes? o Enzymes that are active in acidic conditions What pumps H+ ions into the Lysosome to make it acidic enough for enzymes to become active? o V-Type ATPase Why does the acidic environment inside not destroy the Lysosomal membranes? o They are heavily glycosylated What is a multi-vesicular body? How do you know which receptors need to be degraded? o Multivesicular bodies engulf membrane proteins that receive a monoubiquitin tag (for degradation). They engulf the portion of the membrane so a lysosome can fuse and degrade the contents. o This is a down regulation of membrane receptors o It is an intermediate between an Early Endosome and a Late Endosome What is Autophagy? How does it work? o Autophagy is when the cell envelops and degrades its own organelles. o It does this by the ER engulfing the old organelle and then fusing with a lysosome so that it can be degraded. What is a Lipofuscin? o It is a residual body that does not expel its indigestible contents from the cell, so it remains as a pigmented granule. LYSOSOMAL STORAGE DISEASES Are Lysosomal Storage Diseases dominant or recessive? o Recessive. What is the exception to the mode of inheritance pattern? o Hunter’s – X-Linked I-Cell Disease: What is this disease categorized under? o Mucolipidosis What is deficient? o N-acetylglucosamine phosphotransferase enzyme What does this cause molecularly? o The M6P tag is not added to the enzymes that should be going to the lysosome. Because they do not have this tag, they are secreted extracellularly. The lysosome then has no enzymes to break down ANYTHING, leading to an accumulation of undigested substrates. What are some symptoms of I-Cell Disease? o Skeletal Abnormalities o Coarse Facial Features o Life Expectancy – Less than 10 years Pseudo-Hurler Polydystrophy What is this diseased characterized as? o Mucolipidosis What is it? o A milder form of I-Cell Disease What is the difference in patient presentation? o Because some enzymes get the M6P tag and go into the lysosome: Later onset of symptoms Survival into adulthood Hurler Syndrome What is this disease characterized as? o Mucopolysaccharidoses What is deficient? o A-L-iduronidase enzyme What accumulates in the lysosome? o Dermantan Sulphate and Heparan Sulphate What symptoms are specific to Hurler Disease? o Hirsutism (hair on the face) o Corneal Clouding What is the severity? o Most severe Scheie & Hurler-Scheie Syndrome What is this disease characterized as? o Mucopolysaccharidoses What is deficient? o A-L-iduronidase Enzyme What is the severity? o It is a milder form of Hurler Syndrome Hunter Syndrome What is this disease characterized as? o Mucopolysaccharidoses What is deficient? o Iduronodate Sulphatase What accumulates in the lysosome? o Dermatan Sulphate and Heparan Sulphate What is unique about the Mode of Inheritance? o It is X-Linked What is different about the symptoms? o NO Corneal Clouding Sanfilippo Syndrome What is the disease characterized as? o Mucopolysaccharidoses What is the defect in? o Heparan Sulphate Degradation What is unique about the symptoms? o It isn’t as severe as Hurler because Sanfilipo patients live longer, but they have the most severe behavioral problems Morquio Syndrome What is the disease characterized as? o Mucopolysaccharidoses What is the defect in? o Keratan Sulphate Degradation What is the unique symptom? o Pigeon Chest o Normal IQ What is it often confused with? o Skeletal Dysplasia Metachromatic Leukodystrophy What is the deficiency in? o ARSA (Arylsulfatase A) What is the cause of the deficient enzyme? o Defective degradation of sulfatides = destruction of myelin How does the patient present? o Paralysis and Cognitive Decline Chediak-Higashi Syndrome What is the deficiency in? o CHS1/LYST What is the cause of the deficient enzyme? o Lysosomal Trafficking Regulatory Protein What happens as a result? o Delayed fusion of phagosome with lysosome o Autophagocytosis of melanosomes in melanocytes albinism o Defects in NK Cells and Platelets What is the effect of the mutation on USMLE? o Defect in microtubule polymerization