Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Similar Sequence Similar Function
Charles Yan
Spring 2006
From Sequence to Function
Protein sequence determine protein function. Thus
similar protein sequences have similar functions
One approach to predict function for a new protein is to
search for similar proteins (homologues) whose functions
are known. If the similarities are high, it is likely that the
new protein has the same functions as its homologues
2
Homologue Search
Basic Local Alignment Search Tool (BLAST) finds regions
of local similarity between sequences. The program
compares nucleotide or protein sequences to sequence
databases and calculates the statistical significance of
matches. BLAST can be used to infer functional and
evolutionary relationships between sequences as well as
help identify members of gene families
3
Dynamic Programming
a1a2a3…am
b1b2b3…bn
Mi,j = MAX {
}
Mi-1, j-1 + Si,j (match/mismatch)
Mi,j-1 + w (gap in sequence #1)
Mi-1,j + w (gap in sequence #2)
4
Dynamic Programming
G A A T T C A G T T A (sequence #1)
G G A T C G A (sequence #2)
Si,j = 1 (match)
Si,j = 0 (mismatch score)
w = 0 (gap penalty)
5
Dynamic Programming
M1,1 = MAX[M0,0 + 1, M1, 0 + 0, M0,1 + 0] = MAX [1, 0, 0] = 1
6
Dynamic Programming
7
Dynamic Programming
8
Global and Local Alignment
A global alignment is an optimal alignment that includes
all characters from each sequence, whereas a local
alignment is an optimal alignment that includes only
the most similar local region or regions.
9
BLAST
The BLAST programs (Basic Local Alignment Search
Tools) are a set of sequence comparison algorithms
introduced in 1990 that are used to search sequence
databases for optimal local alignments to a query.
Break the query and database sequences into fragments
("words"), and initially seek matches between fragments.
The initial search is done for a word of length "W" that
scores at least "T" when compared to the query using a
given substitution matrix.
Word hits are then extended in either direction in an attempt
to generate an alignment with a score exceeding the
threshold of "S". The "T" parameter dictates the speed and
sensitivity of the search.
10
11
12
13
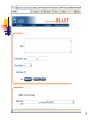
BLAST
Web interface: http://www.ncbi.nlm.nih.gov/BLAST/
Download
http://www.ncbi.nlm.nih.gov/BLAST/download.shtml
14
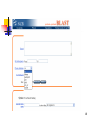
BLAST
15
BLAST
16
17
18
19
BLAST
20
Substitution Matrix
A substitution matrix containing
values proportional to the
probability that amino acid i
mutates into amino acid j for all
pairs of amino acids
21
Substitution Matrix
The BLOSUM family
BLOSUM matrices are based on local alignments.
BLOSUM 62 is a matrix calculated from comparisons of
sequences with no less than 62% divergence.
All BLOSUM matrices are based on observed
alignments; they are not extrapolated from
comparisons of closely related proteins.
BLOSUM 62 is the default matrix in BLAST 2.0. Though
it is tailored for comparisons of moderately distant
proteins, it performs well in detecting closer
relationships. A search for distant relatives may be
more sensitive with a different matrix.
22
Substitution Matrix
The PAM family
PAM matrices are based on global alignments of
closely related proteins.
The PAM1 is the matrix calculated from comparisons
of sequences with no more than 1% divergence.
Other PAM matrices are extrapolated from PAM1.
23
Substitution Matrix
The relationship between BLOSUM and PAM
substitution matrices. BLOSUM matrices with
higher numbers and PAM matrices with low numbers
are both designed for comparisons of closely related
sequences. BLOSUM matrices with low numbers and
PAM matrices with high numbers are designed for
comparisons of distantly related proteins. If distant
relatives of the query sequence are specifically being
sought, the matrix can be tailored to that type of
search.
24
25
Raw Score S
The raw score S for an alignment is calculated by
summing the scores for each aligned position and the
scores for gaps
26
Bit Score S'
Raw scores have little meaning without detailed knowledge
of the scoring system used, or more simply its statistical
parameters K and lambda. Unless the scoring system is
understood, citing a raw score alone is like citing a
distance without specifying feet, meters, or light years.
By normalizing a raw score using the formula
one attains a "bit score" S', which has a standard set of
units.
27
Bit Score S'
The value S' is derived from the raw alignment score S in
which the statistical properties of the scoring system
used have been taken into account. Because bit scores
have been normalized with respect to the scoring
system, they can be used to compare alignment scores
from different searches.
28
Significance
The significance of each alignment is computed as a P value or an E
value
E value: Expectation value. The number of different alignents with
scores equivalent to or better than S that are expected to occur in
a database search by chance. The lower the E value, the more
significant the score.
P value :The probability of an alignment occurring with the score
in question or better. The p value is calculated by relating the
observed alignment score, S, to the expected distribution of HSP
scores from comparisons of random sequences of the same length
and composition as the query to the database. The most highly
significant P values will be those close to 0. P values and E values
are different ways of representing the significance of the
alignment.
29
E-value
In the limit of sufficiently large sequence lengths m and n, the
statistics of HSP scores are characterized by two parameters, K and
lambda. Most simply, the expected number of HSPs with score at
least S is given by the formula
We call this the E-value for the score S.
This formula makes eminently intuitive sense. Doubling the length
of either sequence should double the number of HSPs attaining a
given score. Also, for an HSP to attain the score 2x it must attain the
score x twice in a row, so one expects E to decrease exponentially
with score. The parameters K and lambda can be thought of simply
as natural scales for the search space size and the scoring system
respectively.
30
P-value
The number of random HSPs with score >= S is described by a Poisson
distribution. This means that the probability of finding exactly a HSPs
with score >=S is given by
where E is the E-value of S given by equation (1) above. Specifically
the chance of finding zero HSPs with score >=S is e-E, so the
probability of finding at least one such HSP is
This is the P-value associated with the score S. For example, if one
expects to find three HSPs with score >= S, the probability of finding
at least one is 0.95. The BLAST programs report E-value rather than Pvalues because it is easier to understand the difference between, for
example, E-value of 5 and 10 than P-values of 0.993 and 0.99995.
31
32
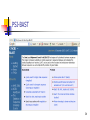
PSI-BAST
Position specific iterative BLAST (PSI-BLAST) refers to a
feature of BLAST 2.0 in which a profile (or position
specific scoring matrix, PSSM) is constructed
(automatically) from a multiple alignment of the highest
scoring hits in an initial BLAST search. The PSSM is
generated by calculating position-specific scores for each
position in the alignment. Highly conserved positions
receive high scores and weakly conserved positions
receive scores near zero. The profile is used to perform a
second (etc.) BLAST search and the results of each
"iteration" used to refine the profile. This iterative
searching strategy results in increased sensitivity.
PSI-BLAST uses the blastp program exclusively, so there is
no need to select the program.
33
PSI-BAST
34
PSI-BAST
The threshold value for inclusion in the position specific
matrix used for PSI-BLAST iterations. Hits with Evalue less than this threshold will be used to
constructed the for next round.
35
PSI-BAST
36
PHI-BLAST
PHI-BLAST (Pattern-Hit Initiated BLAST) is a search
program that combines matching of regular expressions
with local alignments surrounding the match. Given a
protein sequence S and a regular expression pattern P
occurring in S, PHI-BLAST helps answer the question: What
other protein sequences both contain an occurrence of P
and are homologous to S in the vicinity of the pattern
occurrences? PHI-BLAST may be preferable to just
searching for pattern occurrences because it filters out
those cases where the pattern occurrence is probably
random and not indicative of homology.
37
PHI-BLAST
38