Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Particle in a box wikipedia , lookup
Density functional theory wikipedia , lookup
Molecular orbital wikipedia , lookup
Atomic theory wikipedia , lookup
Atomic orbital wikipedia , lookup
Hartree–Fock method wikipedia , lookup
Theoretical and experimental justification for the Schrödinger equation wikipedia , lookup
Franck–Condon principle wikipedia , lookup
X-ray fluorescence wikipedia , lookup
X-ray photoelectron spectroscopy wikipedia , lookup
Tight binding wikipedia , lookup
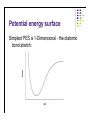
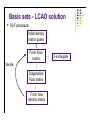
Molecular Quantum Chemistry Dimensionality l l l How many dimensions are there, formally? To define an atom’s location in 3-dimensional space in principle takes 3 coordinates (e.g., x, y, and z in some laboratory reference frame) But, the problem should not depend on the absolute location of the atoms, only on their location relative to one another (i.e., the molecular geometry) So, a typical system has the same dimensionality as the number of degrees of freedom of the molecule or system (3N-6 where N>2 is the number of atoms). Potential energy surface l l l l Normally used for single molecules - for collections of molecules, thermodynamics comes into play, although people still think in terms of PES. Captures the idea that each structure - that is, geometry - has associated with it a unique energy Since geometry changes are smooth, this creates a smooth energy “landscape” Chemistry becomes topology... Potential energy surface Simplest PES is 1-Dimensional - the diatomic bond stretch: Potential energy surface For more dimensions we take “slices”: From Cramer, Essentials of Computational Chemistry Potential energy surface A reaction pathway is a 1-D slice of a PES. ABCA+BC pathway: From Cramer, Essentials of Computational Chemistry Potential energy surface l l l l l Based on the Born-Oppenheimer approximation Inherently classical with respect to the nuclei (but quantum in the electronics) Does not show kinetic energy! Total energy (PE + KE) is conserved at a constant value. At T = 0 K, our molecule will want to be at the lowest possible potential energy, i.e., at a minimum on the PES. Finding ways of locate the minima efficiently is essential in computational chemistry Between any two minima (valley bottoms) the lowest energy path will pass through a maximum at a saddle point. In chemistry, we call that saddle point a transition-state structure. Potential energy surface l l l From Lewars, Computational Chemistry Features: minima, saddle points; How do we describe these mathematically? Reaction coordinates are usually not one of the normal coordinates, but composites. Potential energy surface Fundamental points: l l l It’s clear the PES is useful, so how can I construct it for an arbitrary system (defined simply by the molecular formula)? It seems that for equilibrium and rate constants, I don’t actually need to know the whole surface, only the energies of critical points (minima and saddle points) - is there a way to find these without mapping out the entire surface in detail? If I don’t do the whole surface, can I be sure that I know about the locations of all the critical points and how they relate to one another? What about the KE? l l l l Remember that we are only considering nuclei for the PES. It is intrinsically related to temperature. At 0K, only vibrational kinetic energy. In more accurate calculations, ZPE should be added to the PES. We need to go to statistical mechanics to truly incorporate temperature! Basis sets - LCAO solution l The form of the Fock matrix elements is: Density matrix elements (the total electron density in the overlap region of θl and θm) Basis sets - LCAO solution l SCF procedure: Initial density matrix guess Form Fock matrix iterate Diagonalize Fock matrix Form new density matrix 2-e integrals HF - SCF solution Points to note: l Fock operator depends on occupied MO’s only; l The procedure produces Nbasis total MO’s; l There are Nbasis-Nelec (or Nbasis-Nelec/2) unoccupied MO’s; l These have no direct physical interpretation except as electron affinities. There are Nbasis4 two-electron integrals to solve - this is how the method scales with problem size. This can be reduced in actual calculations trough a number of tricks usually down to Nbasis2. l l l l Other basis sets may be used (like Gaussians or multiple zeta STO’s). More about this in the basis sets section. As basis set size is increases, a lower energy is obtained (as per the variational principle). This is never the exact solution of the Sch. equation. Hartree-Fock l l l For a molecule with n electrons, n spinorbitals are obtained. Int(n/2) spatial orbitals are occupied (occupied orbitals), n-Int(n/2) are unoccupied (virtual orbitals). If n is even and all the electrons are paired up (closed shell), one can assume that the spatial components are the same for a pair of electrons in the same orbital (a restricted Hartree-Fock wavefn): In this case, one only needs to find n/2 spatial orbitals through the HF procedure. Hartree-Fock l l Open shells occur when there are electrons that are not paired up: when n is even but there is a triplet state (2S +1=3) or when n is odd (a doublet: 2S+1=2) In the restricted open shell formalism, all spatial orbitals for paired-up electrons are considered to be identical. For Li, l Often for a chemical problem a better way to go is an unrestricted calculation, where all spinorbitals are calculated independently: l UHF gives a lower energy than RHF, but the problem is that the UHF wavefunction is not an eigenfunction of the spin operator S2: spin is undefined for a UHF wavefunction (this is called the spin contamination problem).