Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
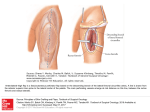
Biochimica et Biophysica Acta 1736 (2005) 30 – 37 http://www.elsevier.com/locate/bba Review The 5-lipoxygenase pathway in arterial wall biology and atherosclerosis Katharina Lötzer a,1, Colin D. Funk b, Andreas J.R. Habenicht a,*,1 a Institute for Vascular Medicine, Friedrich-Schiller-University of Jena, Bachstr. 18, 07743 Jena, Germany Departments of Physiology and Biochemistry, Queen’s University, Kingston, Ontario, Canada K7L 3N6 b Received 2 June 2005; received in revised form 5 July 2005; accepted 8 July 2005 Available online 21 July 2005 Abstract Leukotrienes (LTs) are powerful inflammatory lipid mediators derived from the 5-lipoxygenase (5-LO) cascade of arachidonic acid. Recent clinical, population genetic, cell biological, and mouse studies indicate participation of the 5-LO pathway in atherogenesis and arterial wall remodeling. 5-LO is expressed by leukocytes including blood monocytes, tissue macrophages, dendritic cells, neutrophils, and mast cells. LTB4 and the cysteinyl LTs LTC4, LTD4, and LTE4, act through two BLT and two cysLT receptors that are differentially expressed on hematopoietic and arterial wall cells. The precise roles of LTs or the LT receptors in cardiovascular physiology remain largely to be explored. In this review, we will discuss what is currently known about the 5-LO atherosclerosis connection. We will attempt to propose strategies to further explore potential links between the 5-LO pathway and blood vessel physiology and disease progression. D 2005 Elsevier B.V. All rights reserved. Keywords: 5-Lipoxygenase; Leukotriene; Arachidonic acid; Inflammation; Cardiovascular disease; Atherogenesis 1. The 5-LO pathway of arachidonic acid metabolism produces mediators of blood vessel homeostasis and inflammation LTs, i.e., LTB4 and the cysLTs (i.e. LTC4, LTD4, and LTE4 also known as ‘‘slow reacting substances of anaphylaxis’’), constitute a group of 5-LO-derived arachidonic acid oxidation products that exert multiple activities in the cardiovascular (CV) system [1 – 3]. LT formation is initiated by activation of cytosolic phospholipase A2 resulting in arachidonic acid release from membrane glycerophospholipids [3]. Metabolism of unesterified arachidonic acid to LTs is achieved by 5-LO together with an associated protein, referred to as 5-LO-activating protein (FLAP) that appears to serve as an arachidonic acid binding and transfer protein thereby facilitating 5-LO enzyme activity. The intermediate metabolite, i.e. LTA4, is converted either to LTB4 or to cysLTs by the downstream enzymes LTA4 hydrolase and * Corresponding author. E-mail address: [email protected] (A.J.R. Habenicht). 1 www.med.uni-jena.de/ivm/. 1388-1981/$ - see front matter D 2005 Elsevier B.V. All rights reserved. doi:10.1016/j.bbalip.2005.07.001 LTC4 synthase, respectively [3]. LTs exert biological effects through binding to and activation of one of four G-proteincoupled putative 7 membrane domain-type cell surface receptors (see below). Expression patterns of LT receptors on target cells in blood vessels and atherosclerotic lesions are likely to govern the biological responses in vivo. There are few leukocytes in normal arteries most of which are found in the loose connective tissue that surrounds blood vessels, i.e., the lamina adventitia. In human diseased arteries, the number of 5-LO+ macrophages expands during lesion formation in patients afflicted with late stage atherosclerosis. In clinically significant atherosclerosis, macrophages make up a significant portion of plaques. It seems likely that these macrophages produce LTs although physiological agonists that trigger arachidonic acid liberation and LT biosynthesis in vivo have not yet been identified [4]. We proposed that LTs may trigger circuits of arterial wall inflammation and remodeling during clinically significant stages of atherosclerosis and have referred to this 5-LO involvement in arterial wall biology as the 5-LO atherosclerosis hypothesis (Fig. 1). Of leukocytes that are found in atherosclerotic lesions, macrophages, foam cells, dendritic cells, and mast cells express 5-LO, whereas K. Lötzer et al. / Biochimica et Biophysica Acta 1736 (2005) 30 – 37 31 Fig. 1. The 5-LO atherosclerosis hypothesis of arterial wall inflammation. Macrophages/foam cells/dendritic cells express cytosolic phospholipase A2 (cPLA2) and the 5-LO cascade. cPLA2 activation hydrolyzes arachidonic acid (AA) from the sn2 position of membrane glycerophospholipids. The released unesterified AA binds to FLAP which transfers it to 5-LO facilitating AA conversion to 5-H(P)ETE (5-hydro(pero)xyeicosatetraenoic acid) and LTA4. LTA4 serves as substrate for LTC4 synthase to generate LTC4/LTD4 (black arrows) or for LTA4 hydrolase to generate LTB4 (red arrows). LT formation may occur in lamina intima lesions or in the lamina adventitia. Macrophages, T cells, mast cells, SMCs, and endothelial cells express LT-Rs. CysLTs act on cysLT1-Rs on macrophages in an autocrine (bent black arrows) or on cysLT1-R on other neighboring cells including lymphocytes and/or SMCs in a paracrine fashion (straight black arrows). CysLTs may also activate endothelial cells in a paracrine manner (straight black arrows). Moreover, LTB4 may act on neighboring cells including, SMCs, and T lymphocytes in a paracrine fashion (red straight arrows) and on macrophages in an autocrine fashion (red bent arrows). Inset shows an endothelial cell and genes that have been identified by microarray analyses 60 min after stimulation of cultured human umbilical vein endothelial cells with 100 nM LTD4: CAMs (cell adhesion molecules), MIP-2 (macrophage inflammatory protein 2), IL-8 (interleukin 8). Note that the precise LT-R pattern on cells forming inflammatory circuits in the arterial wall has not yet been determined. T cells do not. However, all plaque-forming cells including T cells, endothelial cells, and smooth muscle cells may express LT receptors. Lipid mediators with prominent proinflammatory activities such as cholesterol oxidation products, lysophospholipids, ceramide, platelet-activating factor, oxidized long chain unsaturated fatty acids, oxidized LDL, and eicosanoids have long been proposed to contribute to arterial wall inflammation [5] but the precise role of each largely remains to be determined. Though recognized more than 20 years ago by Corey and coworkers for their effects on intact blood vessels [6], LTs have not been perceived as promoters of CV disease (CVD). Evidence regarding a role of the 5-LO pathway in atherogenesis has only recently emerged from past in vitro [7 –14], morphological [4], and pharmacological studies [15 – 20] and data from genetically engineered mice [21,22] (see below). Most in vivo effects of added LTs involve interaction of leukocytes and the endothelium [17]. Actions of LTs in blood vessels include coronary artery contraction [15,18], decline in left ventricular contractility [18], edema formation [17], blood pressure regulation, and leukocyte recruitment into the perivascular space [17]. LTs promote P-selectin surface expression, von Willebrand factor secretion, and platelet-activating factor synthesis in cultured endothelial cells [7,11,13], and stimulate arterial smooth muscle cell proliferation in vitro [12]. Moreover, LT-dependent actions on immune cells could conceivably be relevant for the biology of the arterial wall [23 –25]. The 5-LO cascade is strongly expressed in activated macrophages and dendritic cells, LTs activate T and B cells in vitro, appear to affect skin Langerhans cell migration to regional lymph nodes in mice [26], and most cells that participate in immune reactions express LT receptors. Therefore, LTs may mediate multiple effects on CV homeostasis. In a clinical study of carotid and coronary artery, and aorta specimens we observed that 5-LO was highly expressed in arterial walls of patients afflicted with clinically significant atherosclerosis [4]. The enzyme localized to macrophages, dendritic cells, and foam cells and the number of 5-LO expressing lesion leukocytes markedly increased during disease progression. These data support a model of atherogenesis in which 5-LO cascadedependent inflammatory circuits made up by several types of leukocytes, smooth muscle cells, and endothelial cells evolve within the blood vessel wall during late stages of lesion development. According to this model, macrophages produce LTs which act on neighboring cells to activate their LT receptors or on the LT receptors of the macrophages themselves implicating both autocrine and paracrine actions of LTs (Fig. 1). The 5-LO atherosclerosis hypothesis is supported by significant expression of all constituents of the 32 K. Lötzer et al. / Biochimica et Biophysica Acta 1736 (2005) 30 – 37 5-LO pathway and the four LT receptors in human diseased arteries. One of several target tissues of endogenously produced LTs in the blood vessel wall may be the endothelium due to its expression of cysLT2 receptors (see below). In mice, LTB4 was shown to trigger T cell recruitment into inflamed tissues [23 – 25]. These data may indicate that both LTB4 and cysLTs participate in blood vessel biology. Several questions, however, remain to be answered: Which cells present in lesions predominantly form cysLTs and which predominantly form LTB4? Answers to this question may be relevant to understand the role of LTs in CV pathophysiology as cysLTs and LTB4 exert very different activities in blood vessels and each LT receptor may transmit distinct signaling pathways to modulate the phenotypes of plaque forming cells (see below). 5-LO+ leukocytes may act from within the lesions and/or from the adventitial connective tissue in the proximity of cells that express LT receptors during late stage atherogenesis [4,27]. As 5-LO+ macrophages/dendritic cells/foam cells localize in the proximity of neoangiogenic blood vessels in areas of inflammation and in the neighborhood of activated T cells [4], they may also establish immune response circuits (Fig. 1). Thus, the formation of 5-LO+ cell infiltrates observed in clinical samples at predilection sites of atherosclerosis, i.e., coronary and carotid arteries and the aorta, is compatible with participation of LTs to endothelial cell dysfunction, intimal edema, leukocyte infiltration, aberrant contractility, smooth muscle cell proliferation, and immune reactivity (Fig. 2). 2. LT receptors may mediate distinct biological responses in diseased arteries LTs act through four G protein-coupled 7 membrane domain-type receptors that appear to be differentially expressed in normal arterial walls and cells forming Fig. 2. 5-LO in human atherosclerotic cardiovascular tissues. (A) Immunohistochemical staining of 5-LO+ cells (red) in type II aorta lesion according to American Heart Association nomenclature, nuclei are labeled by hematoxylin (blue). (B) Immunofluorescence staining of 5-LO+ cells (Cy3, red) next to a small neoangiogenic blood vessel in an intimal type V lesion of a carotid artery. Endothelial cells were labeled using antisera to von Willebrand factor (Cy2, green), DNA was stained with DAPI. atherosclerotic plaques. Two BLT receptors [28,29] and two cysLT receptors [30,31] have been molecularly characterized and transcripts of all 4 LT receptors have been found in diseased human arteries [4]. There appears to be a dichotomy of cysLT receptor expression in blood vessels and hematopoietic cells: Data indicate preferential expression of cysLT1 receptor in monocyte/macrophages and T cells and of cysLT2 receptor in endothelial cells [32]. In addition, though little is known about BLT receptors in the human normal and diseased arterial wall, BLT1 receptors were up-regulated in mouse T cell effector cells (cytotoxic CD8+ effectors, CD8+ central memory cells) and shown to participate in the recruitment of these T cell effectors to inflamed tissues in experimental models [23 –25]. 2.1. LT-induced gene activation CysLTs were found to induce sustained Ca++ transients in human umbilical vein endothelial cells in vitro indicating that LTs are potent endothelial cell activators. We observed activation of extracellular signal-regulated kinases 1 and 2 with a maximum at 10 min following cysLT2 receptor activation (Lötzer and Habenicht, unpublished). To identify the genetic program induced by cysLT2 receptor-dependent signaling in endothelial cells and by cysLT1 receptor activation of monocytes/macrophages, microarray expression analyses were carried out in human umbilical vein endothelial cells and monocytes (MonoMac6 cells) after addition of LTD4 [27]. These data revealed that the LTD4-triggered gene activation programs in the two cell types are very different with only few overlapping genes: Prominently LTD4-triggered genes in endothelial cells were macrophage-inflammatory protein 2 (MIP-2), interleukin 8 (IL-8), transcription factor early growth response 1 and several additional transcription factors (manuscript in preparation) (Inset in Fig. 1). These chemokines and their chemokine receptors may exacerbate CV inflammation by recruiting leukocytes into the arterial wall. By contrast, in human and mouse monocytic cell lines, LTD4 did not affect MIP-2 but induced MIP1a, a T cell chemoattractant [27]. Whereas LTD4’s action in monocytic cells was inhibitable by the cysLT1 receptor antagonist, montelukast, its effect on endothelial cells was not. These data suggested that LTD4 triggers different responses in an LT receptor and/or target cell-specific manner. Moreover, a recent study on the effects of LTB4 in cultured monocytes demonstrated that BLT1 receptor activation mediated induction of a proinflammatory chemokine, monocyte chemoattractant protein-1 [33]. Taken together, these data indicate that the 5-LO pathway may act on plaque forming cells and suggest novel paradigms of CVD inflammation, i.e., the amplification of leukocyte trafficking and differentiation by the 5-LO cascade through induction of peptide chemokines. Evidence indicates that both BLT and cysLT receptors may be involved. K. Lötzer et al. / Biochimica et Biophysica Acta 1736 (2005) 30 – 37 2.2. CV Effects in CysLT2-R knockout and transgenic mice In addition to gene activation, LTs may trigger acute CV effects independent of transcript regulation. This view is supported by recent studies using cysLT2 receptor-deficient [34] and cysLT2 receptor transgenic mice [35]. When the cysLT2 receptor was deleted in mice the resulting phenotype showed decreased vascular permeability and attenuated bleomycin-induced lung fibrosis [34]. The latter finding may also be relevant to vascular fibroproliferative responses similar to that observed during atherosclerotic plaque formation and arterial wall remodeling [36]. In the other mouse study, the cysLT2 receptor was overexpressed in endothelial cells. The resulting phenotype also suggested a role of cysLT2 receptor in blood vessel permeability and indicated that it participates in blood pressure regulation [35]. These mouse models should now allow characterization of additional cysLT2 receptor-mediated events in CV responses to evaluate the potential impact of cysLTs in vivo. While these recent studies indicate a special role of this LT receptor subtype in CV homeostasis, the potential interactions of the 5-LO cascade with the arterial wall may be complex. Further work is required to examine the actions of LTs on each of the cells that participate in atherogenesis. 33 cell phenotype. That a subpopulation of intima lesion macrophage/foam cells of human atherosclerotic lesions of coronary arteries and carotid arteries express dendritic/ Langerhans cell markers further indicates that a subpopulation of lesion macrophages are dendritic cells capable of presenting antigen to naive T cells thereby promoting macrophage/dendritic T cell interactions within the arterial wall [4]. When apoE / mice were fed an atherogenic diet containing cholesterol, sucrose, and cholic acid, aneurysms develop in the subdiaphragmatic area of the abdominal aorta [27,40]. ApoE / /5-LO / mice showed greatly reduced aortic aneurysm incidence. These data are consistent with the view that the 5-LO cascade may affect arterial wall remodeling. We also assayed biomarkers of inflammation in the circulation and observed that MIP1a was strongly increased in all hyperlipidemic mice. Interestingly, 5-LO deletion reduced the extent of MIP1a up-regulation in a gene dose-dependent fashion. These results indicated that hyperlipidemia triggers, by unknown mechanisms, circulating MIP1a levels and that 5-LO interrupts this up-regulation. These data support hitherto unrecognized links between 5-LO, hyperlipidemia, and inflammatory chemokine production in vivo. However, further work is required to elucidate the underlying mechanisms of these associations. 3. The 5-LO pathway in hyperlipidemic mice A locus on mouse chromosome 6, where the 5-LO gene is located, has been demonstrated to confer resistance to atherogenesis in the atherosclerosis-resistant strain CAST/Ei despite hyperlipidemia [37]. It was hypothesized that 5-LO may represent a pro-atherogenic susceptibility gene and data in LDL receptor / mice supported this hypothesis [38]. In addition, pharmacological studies supported a role of LTB4 in the formation of lipid-rich lesions of the aorta in hyperlipidemic mice [39]. We generated LDL / /5-LO / and apoE / /5-LO / mice and examined lesion formation, aneurysm formation, and wire-induced carotid artery responses to injury under a variety of dietary conditions but failed to observe effects of 5-LO deletion in lesion formation and intimal proliferation in response to injury [27] (Nicole John and A.J.R. Habenicht, unpublished). The reasons for these differences may relate to different mouse colonies, immune status, and housing and feeding conditions. When we examined the abdominal aortas of hyperlipidemic mice (apoE / ) for 5-LO expression in leukocytes we observed the presence of two monocyte/macrophage/foam cell populations: 5-LO+ macrophages accumulate in the adventitia and 5-LO macrophages accumulate in lipidrich lesions. We noted that intimal macrophages were CD11c+, whereas adventitial macrophages were CD11cdim. CD11c is a marker of activated macrophages and of dendritic cells. These data are consistent with the possibility that lesion macrophages acquire a dendritic 4. Population genetic studies indicate a role of LTs in atherogenesis The Los Angeles Atherosclerosis Study group reported on patients carrying 5-LO promoter polymorphisms and related them to CVD [41]. The rationale behind the study stems from previously described polymorphisms in the 5LO promoter that had been correlated with 5-LO inhibitor responsiveness in asthma patients and had been presumed to represent loss of function mutations [42]. Carriers of two variant alleles exhibited significantly increased atherosclerosis as assessed by sonographic determination of the intima/media thickness of the common carotid artery. Moreover, intake of N3 polyunsaturated fatty acids (eicosapentaenoic and docosahexaenoic acid) blunted the effect in the 5-LO gene variant carriers whereas dietary intake of the N6 polyunsaturated fatty acids (arachidonic acid and its precursor linoleic acid) enhanced the effect. The data lend support to the hypothesis that these fatty acids compete as substrates for the 5-LO enzyme reaction in vivo and thereby shift the balance of pro- versus anti-inflammatory 5-LOderived eicosanoids. When taken together, these data suggest links between the 5-LO pathway and atherogenesis and between 5-LO and dietary fatty acid intake. However, we agree with the authors in their cautious interpretation of these data pointing out several caveats: there was only a small number of patients carrying the respective variant alleles making the epidemiological data difficult to interpret; no biochemical parameters of 5-LO activity were deter- 34 K. Lötzer et al. / Biochimica et Biophysica Acta 1736 (2005) 30 – 37 mined in these patients but rather, functionality was inferred from the earlier pharmacological study on asthma patients [42]: Promoter reporter transfection studies in cultured cells had yielded functional effects of variant alleles but it remained uncertain whether any of these variants is biologically relevant in relation to LT synthesis in vivo [43 –45]. Thus, functionality of 5-LO promoter genotypes with respect to LT formation in relevant leukocyte populations has not yet been clearly demonstrated in asthma or CVD patients. 5. FLAP genotypes correlate with stroke and/or myocardial infarction Further support for a link between the 5-LO cascade and atherogenesis emerged when investigators from deCODE Genetics reported results of a clinical genetic trial of FLAP polymorphisms. They mapped a gene that increases risk of myocardial infarction and stroke to 13q12 –13 containing the FLAP locus [46]. Since FLAP is believed to be required for LT biosynthesis probably by acting as an arachidonic acid binding and transfer protein, FLAP genetic alterations could result in altered LT formation. In a cohort of 779 patients in Iceland, a particular FLAP gene haplotype (HapA) was associated with a 2-fold greater risk of myocardial infarction and stroke while another cohort in the UK carrying another variant haplotype correlated with an increased risk of myocardial infarction but not stroke [46]. Though the authors determined ex vivo LTB 4 formation in neutrophils, LTB4 levels of the haplotype carriers with myocardial infarction when compared to LTB4 levels of their control myocardial infarction counterparts were not different. Hence, similar to the Los Angeles study on 5-LO promoter polymorphisms, a link between FLAP haplotype and LT formation has not yet been achieved. However, inherent difficulties of ex vivo LT synthesis assays that rely on added ionomycin (a rather strong and unphysiological receptor-independent agonist of LT formation) may have prevented detection of small but functionally important differences. To establish functionality of the genotypes, FLAP assays that are sensitive enough to pick up slight changes in agonist-dependent LT synthesis need to be established to be performed in clinical studies. Meanwhile, the deCODE Genetics investigators reported on a Scottish population of 450 patients with ischemic stroke and 710 controls from Aberdeenshire in which genotyping of seven small nucleotide FLAP polymorphisms was conducted [47]. The Scottish carriers had a significantly greater stroke risk when compared to the controls. Recently, Lõhmussaar et al. showed that sequence variants of FLAP (distinct from the HapA variants reported by deCODE Genetics) in 639 stroke patients in Southern Germany showed a weak correlation with stroke whereas the HapA carriers in the German population did not [48]. When taken together, these studies are consistent with but clearly not yet prove connections between genetic alterations of constituents of the 5-LO cascade and CVD. Following this hypothesis, the deCODE Genetics investigators studied 191 patients with the HapA haplotypes or a newly reported nucleotide polymorphism in the LTA4 hydrolase gene that had experienced a myocardial infarction [49]. The study was designed to assess clinical safety and efficacy of three doses of the FLAP inhibitor, DG-031, and to determine the effects of the drug on clinical parameters of CVD risk such as myeloperoxidase and C-reactive protein [49]. DG-031 was found to be safe, reduced LTB4 formation in the neutrophil ex vivo assay by 25%, and also reduced myeloperoxidase by 12%, whereas C-reactive protein was not affected by the drug. deCODE Genetics announced initiation of a large phase III 2 year outcome study of 1500 patients with previous myocardial infarction to test whether FLAP inhibition will prevent recurrent heart attacks. If the upcoming trials to be conducted between 2005 and 2007 confirm the data of the phase IIa trial and indicate beneficial effects on clinical outcome, conditions for larger drug development programs to treat arterial inflammation using anti-LT drugs will be set. 6. The 5-LO and FLAP population studies reveal that the 5-LO inflammation/atherosclerosis connection may be clinically significant: what may be the underlying mechanism? In the 5-LO promoter polymorphism and FLAP haplotype studies, variant allele carriers were presumed to represent loss of function [41] and gain of function [46] phenotypes, respectively, that were assumed to result in a decrease or increase in LT synthesis in vivo, respectively. However, carriers of both 5-LO and FLAP haplotypes had an increased risk to develop carotid artery disease [41] or myocardial infarction/stroke [46] raising a paradox in the genetic trials: In fact, these results shed doubt on a close connection between LT synthesis and CVD. Moreover, the LT synthesis assays did not reveal significant changes in LT biosynthesis in ex vivo isolated neutrophils in the FLAP haplotype carriers (see online Table 7 of reference [46]). Clearly, if LTs were promoters of CVD inflammation and in that attribute pro-atherogenic, one would expect a decrease in CVD in the variant 5-LO allele carriers and an increase in CVD risk in the HapA carriers in the deCODE Genetics study. Thus, both studies have fallen short in establishing alterations of LT synthesis due to genetic alterations. An alternative explanation, contrary to current belief, is that LTs may have anti-atherogenic properties in the arterial wall and a loss of 5-LO function then would result in a decrease in the defense mechanisms that may normally protect the arterial wall. A further interpretation of the 5-LO population studies would be that all variants are associated with gain of function mutations. These apparent inconsistencies and loose ends need to be addressed to link genotypes to LT K. Lötzer et al. / Biochimica et Biophysica Acta 1736 (2005) 30 – 37 synthesis and LT synthesis to atherosclerosis. As pointed out above, unless reliable assays can determine the functionality of 5-LO/FLAP genotypes with respect to LT synthesis in vivo, little can be said about the molecular basis for the haplotype/clinical outcome results. Clearly, the data would be more compelling if assays of LT synthesis that reflect LT actions in vivo could be associated with the genetic alterations in the population studies. Any coherent 5-LO atherosclerosis hypothesis can only emerge after clarification of these important issues. 7. The 5-LO atherosclerosis hypothesis invites further in depth mechanistic studies into the role of LTs in vascular wall inflammation Results of recent human and mouse studies are consistent with a role of the 5-LO cascade in CVD biology and arterial wall remodeling. The body of evidence supporting this hypothesis has increased considerably but largely relies on the biological activities of high concentrations of added LTs to experimental systems and on the putative pro-inflammatory roles of LTs in asthma. The preliminary clinical trials of 5-LO and FLAP gene variations raise the issue of the underlying molecular mechanisms as well as the functionality of the genetic variants of 5-LO cascade constituents. Thus, it will be important to: (i) examine the relationships between genotypes and 5-LO and/or FLAP activities in vivo by developing reliable test systems that allow to determine subtle changes of LT production in patients afflicted with CVD to perform prospective clinical trials, a task that may be difficult to achieve; (ii) test mouse models of CVD and remodeling in various contexts of injury/inflammation; and (iii) evaluate impacts of tissue-specific 5-LO pathway constituents and LT receptor gene deletions in mice; (iv) identify LT-triggered gene signatures in cultured cells such as smooth muscle cells, endothelial cells, and hematopoietic cells to define possible in vivo candidate LTregulated genes. Once associations between genetic alterations and functionality will have been established, pharmacological intervention trials of patients with CVD will help to determine therapeutic potentials of anti-LT drugs. Testing of anti-LT drugs in selected patient groups for drugs, whose safety has been confirmed for the treatment of chronic lung disease (i.e. in patients that often are also afflicted with CVD), is possible now including cysLT1 receptor antagonists and 5-LO inhibitors [50,51]. Additional molecules in the 5-LO cascade deserve attention as potential drug targets: Inhibitors of LTA4 hydrolase will selectively limit LTB4 production and inhibitors of LTC4 synthase will reduce the burden of cysLTs. Finally, development programs for BLT receptor antagonists and cysLT2 receptor antagonists targeting specific effector sites of the 5-LO cascade should be invigorated for animal and subsequent clinical testing. As these approaches are 35 presently leaving the realm of theory, the question regarding the validity of the 5-LO atherosclerosis hypothesis may be answered in the not too distant future. Acknowledgements The manuscript was supported by grants of the Deutsche Forschungsgemeinschaft (Ha 1083/13-3/13-4/14-1/14-2/125/12-6) and the Interdisziplinäres Zentrum für Klinische Forschung Jena (TP4.4; TP4.9) and the National Institutes of Health (HL58464), Canadian Institutes of Health Research (MOP-67146 and MOP-68930). CDF is holder of a Canada Research Chair in Molecular, Cellular and Physiological Medicine. The authors thank Jilly Evans for comments on the manuscript and Rolf Graebner for Fig. 2. They also apologize to their colleagues for the omission of many references due to space restrictions. References [1] A.W. Ford-Hutchinson, Leukotriene B4 in inflammation, Crit. Rev. Immunol. 10 (1990) 1 – 12. [2] C.D. Funk, Prostaglandins and leukotrienes: advances in eicosanoid biology, Science 294 (2001) 1871 – 1875. [3] B. Samuelsson, Leukotrienes: mediators of immediate hypersensitivity reactions and inflammation, Science 220 (1983) 568 – 575. [4] R. Spanbroek, R. Grabner, K. Lotzer, M. Hildner, A. Urbach, K. Ruhling, M.P. Moos, B. Kaiser, T.U. Cohnert, T. Wahlers, A. Zieske, G. Plenz, H. Robenek, P. Salbach, H. Kuhn, O. Radmark, B. Samuelsson, A.J. Habenicht, Expanding expression of the 5-lipoxygenase pathway within the arterial wall during human atherogenesis, Proc. Natl. Acad. Sci. U. S. A. 100 (2003) 1238 – 1243. [5] R. Ross, Atherosclerosis—An inflammatory disease, N. Engl. J. Med. 340 (1999) 115 – 126. [6] J.A. Burke, R. Levi, Z.G. Guo, E.J. Corey, Leukotrienes C4, D4 and E4: effects on human and guinea-pig cardiac preparations in vitro, J. Pharmacol. Exp. Ther. 221 (1982) 235 – 241. [7] Y.H. Datta, M. Romano, B.C. Jacobson, D.E. Golan, C.N. Serhan, B.M. Ewenstein, Peptido-leukotrienes are potent agonists of von Willebrand factor secretion and P-selectin surface expression in human umbilical vein endothelial cells, Circulation 92 (1995) 3304 – 3311. [8] E.B. Friedrich, A.M. Tager, E. Liu, A. Pettersson, C. Owman, L. Munn, A.D. Luster, R.E. Gerszten, Mechanisms of leukotriene B4triggered monocyte adhesion, Arterioscler. Thromb. Vasc. Biol. 23 (2003) 1761 – 1767. [9] M. Heimburger, J.E. Palmblad, Effects of leukotriene C4 and D4, histamine and bradykinin on cytosolic calcium concentrations and adhesiveness of endothelial cells and neutrophils, Clin. Exp. Immunol. 103 (1996) 454 – 460. [10] A.J. Huang, J.E. Manning, T.M. Bandak, M.C. Ratau, K.R. Hanser, S.C. Silverstein, Endothelial cell cytosolic free calcium regulates neutrophil migration across monolayers of endothelial cells, J. Cell Biol. 120 (1993) 1371 – 1380. [11] T.M. McIntyre, G.A. Zimmerman, S.M. Prescott, Leukotrienes C4 and D4 stimulate human endothelial cells to synthesize platelet-activating factor and bind neutrophils, Proc. Natl. Acad. Sci. U. S. A. 83 (1986) 2204 – 2208. [12] L. Palmberg, H.E. Claesson, J. Thyberg, Leukotrienes stimulate initiation of DNA synthesis in cultured arterial smooth muscle cells, J. Cell Sci. 88 (Pt. 2) (1987) 151 – 159. 36 K. Lötzer et al. / Biochimica et Biophysica Acta 1736 (2005) 30 – 37 [13] K.E. Pedersen, B.S. Bochner, B.J. Undem, Cysteinyl leukotrienes induce P-selectin expression in human endothelial cells via a nonCysLT1 receptor-mediated mechanism, J. Pharmacol. Exp. Ther. 281 (1997) 655 – 662. [14] E. Porreca, C. Di Febbo, A. Di Sciullo, D. Angelucci, M. Nasuti, P. Vitullo, M. Reale, P. Conti, F. Cuccurullo, A. Poggi, Cysteinyl leukotriene D4 induced vascular smooth muscle cell proliferation: a possible role in myointimal hyperplasia, Thromb. Haemost. 76 (1996) 99 – 104. [15] S. Allen, M. Dashwood, K. Morrison, M. Yacoub, Differential leukotriene constrictor responses in human atherosclerotic coronary arteries, Circulation 97 (1998) 2406 – 2413. [16] S.P. Allen, A.H. Chester, P.J. Piper, A.P. Sampson, E.S. Akl, M.H. Yacoub, Effects of leukotrienes C4 and D4 on human isolated saphenous veins, Br. J. Clin. Pharmacol. 34 (1992) 409 – 414. [17] S.E. Dahlen, J. Bjork, P. Hedqvist, K.E. Arfors, S. Hammarstrom, J.A. Lindgren, B. Samuelsson, Leukotrienes promote plasma leakage and leukocyte adhesion in postcapillary venules: in vivo effects with relevance to the acute inflammatory response, Proc. Natl. Acad. Sci. U. S. A. 78 (1981) 3887 – 3891. [18] F. Michelassi, L. Landa, R.D. Hill, E. Lowenstein, W.D. Watkins, A.J. Petkau, W.M. Zapol, Leukotriene D4: a potent coronary artery vasoconstrictor associated with impaired ventricular contraction, Science 217 (1982) 841 – 843. [19] R.J. Secrest, B.M. Chapnick, Endothelial-dependent relaxation induced by leukotrienes C4, D4, and E4 in isolated canine arteries, Circ. Res. 62 (1988) 983 – 991. [20] G. Smedegard, P. Hedqvist, S.E. Dahlen, B. Revenas, S. Hammarstrom, B. Samuelsson, Leukotriene C4 affects pulmonary and cardiovascular dynamics in monkey, Nature 295 (1982) 327 – 329. [21] X.S. Chen, J.R. Sheller, E.N. Johnson, C.D. Funk, Role of leukotrienes revealed by targeted disruption of the 5-lipoxygenase gene, Nature 372 (1994) 179 – 182. [22] J.L. Goulet, R.C. Griffiths, P. Ruiz, R.B. Mannon, P. Flannery, J.L. Platt, B.H. Koller, T.M. Coffman, Deficiency of 5-lipoxygenase accelerates renal allograft rejection in mice, J. Immunol. 167 (2001) 6631 – 6636. [23] K. Goodarzi, M. Goodarzi, A.M. Tager, A.D. Luster, U.H. von Andrian, Leukotriene B4 and BLT1 control cytotoxic effector T cell recruitment to inflamed tissues, Nat. Immunol. 4 (2003) 965 – 973. [24] V.L. Ott, J.C. Cambier, J. Kappler, P. Marrack, B.J. Swanson, Mast cell-dependent migration of effector CD8+ T cells through production of leukotriene B4, Nat. Immunol. 4 (2003) 974 – 981. [25] A.M. Tager, S.K. Bromley, B.D. Medoff, S.A. Islam, S.D. Bercury, E.B. Friedrich, A.D. Carafone, R.E. Gerszten, A.D. Luster, Leukotriene B4 receptor BLT1 mediates early effector T cell recruitment, Nat. Immunol. 4 (2003) 982 – 990. [26] D.F. Robbiani, R.A. Finch, D. Jager, W.A. Muller, A.C. Sartorelli, G.J. Randolph, The leukotriene C(4) transporter MRP1 regulates CCL19 (MIP-3beta, ELC)-dependent mobilization of dendritic cells to lymph nodes, Cell 103 (2000) 757 – 768. [27] L. Zhao, M.P. Moos, R. Grabner, F. Pedrono, J. Fan, B. Kaiser, N. John, S. Schmidt, R. Spanbroek, K. Lotzer, L. Huang, J. Cui, D.J. Rader, J.F. Evans, A.J. Habenicht, C.D. Funk, The 5-lipoxygenase pathway promotes pathogenesis of hyperlipidemia-dependent aortic aneurysm, Nat. Med. 10 (2004) 966 – 973. [28] T. Yokomizo, T. Izumi, K. Chang, Y. Takuwa, T. Shimizu, A Gprotein-coupled receptor for leukotriene B4 that mediates chemotaxis, Nature 387 (1997) 620 – 624. [29] T. Yokomizo, K. Kato, K. Terawaki, T. Izumi, T. Shimizu, A second leukotriene B(4) receptor, BLT2. A new therapeutic target in inflammation and immunological disorders, J. Exp. Med. 192 (2000) 421 – 432. [30] C.E. Heise, B.F. O’Dowd, D.J. Figueroa, N. Sawyer, T. Nguyen, D.S. Im, R. Stocco, J.N. Bellefeuille, M. Abramovitz, R. Cheng, D.L. Williams Jr., Z. Zeng, Q. Liu, L. Ma, M.K. Clements, N. Coulombe, Y. [31] [32] [33] [34] [35] [36] [37] [38] [39] [40] [41] [42] [43] [44] Liu, C.P. Austin, S.R. George, G.P. O’Neill, K.M. Metters, K.R. Lynch, J.F. Evans, Characterization of the human cysteinyl leukotriene 2 receptor, J. Biol. Chem. 275 (2000) 30531 – 30536. K.R. Lynch, G.P. O’Neill, Q. Liu, D.S. Im, N. Sawyer, K.M. Metters, N. Coulombe, M. Abramovitz, D.J. Figueroa, Z. Zeng, B.M. Connolly, C. Bai, C.P. Austin, A. Chateauneuf, R. Stocco, G.M. Greig, S. Kargman, S.B. Hooks, E. Hosfield, D.L. Williams Jr., A.W. Ford-Hutchinson, C.T. Caskey, J.F. Evans, Characterization of the human cysteinyl leukotriene CysLT1 receptor, Nature 399 (1999) 789 – 793. K. Lotzer, R. Spanbroek, M. Hildner, A. Urbach, R. Heller, E. Bretschneider, H. Galczenski, J.F. Evans, A.J. Habenicht, Differential leukotriene receptor expression and calcium responses in endothelial cells and macrophages indicate 5-lipoxygenase-dependent circuits of inflammation and atherogenesis, Arterioscler. Thromb. Vasc. Biol. 23 (2003) e32 – e36. L. Huang, A. Zhao, F. Wong, J.M. Ayala, M. Struthers, F. Ujjainwalla, S.D. Wright, M.S. Springer, J. Evans, J. Cui, Leukotriene B4 strongly increases monocyte chemoattractant protein-1 in human monocytes, Arterioscler. Thromb. Vasc. Biol. 24 (2004) 1783 – 1788. T.C. Beller, A. Maekawa, D.S. Friend, K.F. Austen, Y. Kanaoka, Targeted gene disruption reveals the role of the cysteinyl leukotriene 2 receptor in increased vascular permeability and in bleomycininduced pulmonary fibrosis in mice, J. Biol. Chem. 279 (2004) 46129 – 46134. Y. Hui, Y. Cheng, I. Smalera, W. Jian, L. Goldhahn, G.A. Fitzgerald, C.D. Funk, Directed vascular expression of human cysteinyl leukotriene 2 receptor modulates endothelial permeability and systemic blood pressure, Circulation 110 (2004) 3360 – 3366. M.R. Ward, G. Pasterkamp, A.C. Yeung, C. Borst, Arterial remodeling. Mechanisms and clinical implications, Circulation 102 (2000) 1186 – 1191. M. Mehrabian, J. Wong, X. Wang, Z. Jiang, W. Shi, A.M. Fogelman, A.J. Lusis, Genetic locus in mice that blocks development of atherosclerosis despite extreme hyperlipidemia, Circ. Res. 89 (2001) 125 – 130. M. Mehrabian, H. Allayee, J. Wong, W. Shi, X.P. Wang, Z. Shaposhnik, C.D. Funk, A.J. Lusis, Identification of 5-lipoxygenase as a major gene contributing to atherosclerosis susceptibility in mice, Circ. Res. 91 (2002) 120 – 126. R.J. Aiello, P.A. Bourassa, S. Lindsey, W. Weng, A. Freeman, H.J. Showell, Leukotriene B4 receptor antagonism reduces monocytic foam cells in mice, Arterioscler. Thromb. Vasc. Biol. 22 (2002) 443 – 449. A. Daugherty, L.A. Cassis, Mouse models of abdominal aortic aneurysms, Arterioscler. Thromb. Vasc. Biol. 24 (2004) 429 – 434. J.H. Dwyer, H. Allayee, K.M. Dwyer, J. Fan, H. Wu, R. Mar, A.J. Lusis, M. Mehrabian, Arachidonate 5-lipoxygenase promoter genotype, dietary arachidonic acid, and atherosclerosis, N. Engl. J. Med. 350 (2004) 29 – 37. J.M. Drazen, C.N. Yandava, L. Dube, N. Szczerback, R. Hippensteel, A. Pillari, E. Israel, N. Schork, E.S. Silverman, D.A. Katz, J. Drajesk, Pharmacogenetic association between ALOX5 promoter genotype and the response to anti-asthma treatment, Nat. Genet. 22 (1999) 168 – 170. K.H. In, K. Asano, D. Beier, J. Grobholz, P.W. Finn, E.K. Silverman, E.S. Silverman, T. Collins, A.R. Fischer, T.P. Keith, K. Serino, S.W. Kim, G.T. De Sanctis, C. Yandava, A. Pillari, P. Rubin, J. Kemp, E. Israel, W. Busse, D. Ledford, J.J. Murray, A. Segal, D. Tinkleman, J.M. Drazen, Naturally occurring mutations in the human 5-lipoxygenase gene promoter that modify transcription factor binding and reporter gene transcription, J. Clin. Invest. 99 (1997) 1130 – 1137. E.S. Silverman, J.M. Drazen, Genetic variations in the 5-lipoxygenase core promoter. Description and functional implications, Am. J. Respir. Crit. Care Med. 161 (2000) S77 – S80. K. Lötzer et al. / Biochimica et Biophysica Acta 1736 (2005) 30 – 37 [45] E.S. Silverman, J. Du, G.T. De Sanctis, O. Radmark, B. Samuelsson, J.M. Drazen, T. Collins, Egr-1 and Sp1 interact functionally with the 5-lipoxygenase promoter and its naturally occurring mutants, Am. J. Respir. Cell Mol. Biol. 19 (1998) 316 – 323. [46] A. Helgadottir, A. Manolescu, G. Thorleifsson, S. Gretarsdottir, H. Jonsdottir, U. Thorsteinsdottir, N.J. Samani, G. Gudmundsson, S.F. Grant, G. Thorgeirsson, S. Sveinbjornsdottir, E.M. Valdimarsson, S.E. Matthiasson, H. Johannsson, O. Gudmundsdottir, M.E. Gurney, J. Sainz, M. Thorhallsdottir, M. Andresdottir, M.L. Frigge, E.J. Topol, A. Kong, V. Gudnason, H. Hakonarson, J.R. Gulcher, K. Stefansson, The gene encoding 5-lipoxygenase activating protein confers risk of myocardial infarction and stroke, Nat. Genet. 36 (2004) 233 – 239. [47] A. Helgadottir, S. Gretarsdottir, D. St Clair, A. Manolescu, J. Cheung, G. Thorleifsson, A. Pasdar, S.F. Grant, L.J. Whalley, H. Hakonarson, U. Thorsteinsdottir, A. Kong, J. Gulcher, K. Stefansson, M.J. MacLeod, Association between the gene encoding 5-lipoxygenaseactivating protein and stroke replicated in a Scottish population, Am. J. Hum. Genet. 76 (2005) 505 – 509. 37 [48] E. Lohmussaar, A. Gschwendtner, J.C. Mueller, T. Org, E. Wichmann, G. Hamann, T. Meitinger, M. Dichgans, ALOX5AP gene and the PDE4D gene in a central European population of stroke patients, Stroke 36 (2005) 731 – 736. [49] H. Hakonarson, S. Thorvaldsson, A. Helgadottir, D. Gudbjartsson, F. Zink, M. Andresdottir, A. Manolescu, D.O. Arnar, K. Andersen, A. Sigurdsson, G. Thorgeirsson, A. Jonsson, U. Agnarsson, H. Bjornsdottir, G. Gottskalksson, A. Einarsson, H. Gudmundsdottir, A.E. Adalsteinsdottir, K. Gudmundsson, K. Kristjansson, T. Hardarson, A. Kristinsson, E.J. Topol, J. Gulcher, A. Kong, M. Gurney, G. Thorgeirsson, K. Stefansson, Effects of a 5-lipoxygenase-activating protein inhibitor on biomarkers associated with risk of myocardial infarction: a randomized trial, JAMA 293 (2005) 2245 – 2256. [50] R. De Caterina, A. Zampolli, From asthma to atherosclerosis-5lipoxygenase, leukotrienes, and inflammation, N. Engl. J. Med. 350 (2004) 4 – 7. [51] M. Mehrabian, H. Allayee, 5-lipoxygenase and atherosclerosis, Curr. Opin. Lipidol. 14 (2003) 447 – 457.