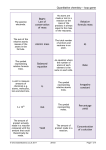
Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
A2 Unit 5 Energetics, Redox and Inorganic Chemistry AQA A2-LEVEL Student Guide to A2 Unit 5 Energetics, Redox and Inorganic Chemistry See me in glorious All programs Shared Areas at: Chemistry Read Mr Lund’s Classes LUND 7 May 2017 1 A2 Chemistry Unit 5 A2 Unit 5 Energetics, Redox and Inorganic Chemistry Enthalpy Changes and Hess’s Law enthalpy changeH = heat exchange at constant pressure (open container) enthalpy change cannot be measured directly but can be determined experimentally H varies with temperature and pressure so Standard Conditions (Ho298) are required: temperature pressure physical state 298 K (25ºC i.e. nominal room temperature) 100kPa don’t put ‘1 atmosphere’ in the exam!!! at room temperature and most stable allotrope (e.g. graphite) the enthalpy of formation of an element is by definition zero here are two enthalpy changes that you learned in AS Chemistry: the enthalpy of is the enthalpy change that occurs when one mole of at 298K and 100 kPa (i.e. standard conditions) formation (Hof,298) a compound is formed from its elements combustion (oc,298) an element or compound is completely combusted in xs oxygen Hess’s Law: the enthalpy change for a reaction is dependent only on the initial and final states of the system and is independent of the route taken. Reactants Products 1 2 Hess’s Law states that 1 = 2 A MARK IN THE EXAM !! Hor,298 = Hof,298 (Products) - Hof,298 (Reactants) Elements Reactants s Products 1 2 Hess’s Law states that 1 = 2 Hor,298 = Hoc,298 (Reactants) - Hoc,298 (Products) Combustion Products LUND 7 May 2017 2 A2 Unit 5 Energetics, Redox and Inorganic Chemistry Enthalpy Definitions note that once again in all these definitions you specify both the quantity and type of the particles initially and finally the enthalpy of is the enthalpy change that occurs when one mole of at 298K and 100 kPa (i.e. standard conditions) atomisation Hoat note the differences between the enthalpies of formation, atomisation and bond dissociation as these are often used incorrectly in calculations (think about the values for Iodine) 1st ionisation energy Hoi(1) gaseous singly charged cations are formed from gaseous atoms revise the trends (periods and groups) of first ionisation energies in addition to the changes in magnitude (shells) and equations for successive ionisation energies of the same element 1st electron affinity Hoea(1) gaseous atoms are formed from an element in its standard state gaseous singly charged anions are formed from gaseous atoms you should be able to write an equation for the gain of an electron by any given atom/ion the first electron affinity is exothermic as the vacancy in the subshell is subject to an attractive force from the nucleus that is not completely screened. the second electron affinity is endothermic since an electron is being moved towards a negative ion and mutual repulsion must be overcome (subsequent additions are increasingly endothermic as the size of the negative charge increases) EXOTHERMIC lattice enthalpy HoL an ionic lattice is formed from its gaseous ions lattice dissociation enthalpy ENDOTHERMIC an ionic compound separates into gaseous ions Note that these have the same numerical value but opposite signs lattice enthalpy is higher if the charges are higher and/or the ions smaller as the electrostatic force of attraction is greater – see 169 (note it’s the same as lattice energy for A-level use) Lattice enthalpy and enthalpy of formation are not the same thing: Lattice enthalpy starts from gaseous ions. Enthalpy of formation starts from elements in their normal states. lattice energy cannot be determined experimentally as it is impossible to react an exact amount of gaseous anions and cations and measure the energy released. LUND 7 May 2017 3 A2 Unit 5 Energetics, Redox and Inorganic Chemistry the value of the lattice energy can be derived theoretically using an electrostatic relationship between charge sizes and distances but this makes two incorrect assumptions: 1 2 experimental values are higher than theoretically calculated values for HoL as there is in reality a degree of covalency in all ionic compounds as the electrostatic attraction of the cations will distort the valence electron cloud of the anions in most cases (small ions that are singly charged) there is a reasonable agreement between experimental and theoretical data (within 5%) i.e. the ionic model is adequate. however, the deviation is more significant i.e. the degree of covalency increased if: 1 2 3 the ions are point sources the ions do not mutually interact such as to distort their shape i.e that they are purely ionic and exhibit no degree of covalency the cations are smaller the anions are larger the charge on the ions is larger a small cation with a charge of 2+ or higher will present a highly polarising field to an anion the effect of which will be more significant if that anion is relatively large the anionic electron clouds will be distorted (polarised) towards the polarising field of the cation (this is not dissimilar to the tenuous outer atmosphere of a red giant star becoming distorted in association with a strong gravitational field) this can lead to an overlap of electron clouds i.e. the compound is in effect covalent Summary Questions A2 Chemistry (Nelson Thornes) AQA Chemguide Page 166 Page 174 1–3 2 163 – 166, 169, 174 Lattice enthalpy, ionic structures s-cool: Chemical Energetics LUND 7 May 2017 4 A2 Unit 5 Energetics, Redox and Inorganic Chemistry Born-Haber Cycles a Born-Haber Cycle for a simple ionic solid can be constructed using Hess’s Law (including ionisation energy, electron affinity, lattice energy, atomisation energy) Note that, unlike the energy cycle diagrams met at AS-level, these are energy level diagrams in that they are drawn with up = endothermic and down = exothermic change elements are referenced to zero NaCl and MgCl2 are shown in your textbook for oxides/sulphides the second electron affinity will be endothermic (i.e. back up) but why not MgCl without a second (endothermic) ionisation energy or MgCl3 where a 3+ ion is forming the lattice giving a much higher (exothermic) lattice energy (see page 170) ionisation energy and lattice energy are the two major contributory values Enthalpies of Solution (Hosol) the enthalpy of is the enthalpy change that occurs when one mole of at 298K and 100 kPa (i.e. standard conditions) solution Hosol solute is completely dissolved in water hydration Hohyd isolated gaseous ions are hydrated hydration involves ion-dipole electrostatic forces hence ionic solids can dissolve in polar solvents such as water the payback of hydration enthalpy is not available in non-polar solvents rendering the process too endothermic overall to be possible solubility is essentially a trade off between overcoming the enthalpy of lattice dissociation enthalpy (endothermic in this direction) and enthalpies of hydration (always exothermic) solubility is feasible if this is an exothermic outcome overall (e.g. NaOH(aq)) although ENTROPY (see later) will also have a say if the enthalpy of solution is only slightly endothermic (e.g. NH4NO3(aq)) Hess’s Law can be used to construct a solvation energy cycle (see 173) Summary Questions Page 172 1 Page 174 1 Pages 171 – 2 and Questions 1 - 4 Page 184 1 Page 248 5, 6 (read ‘entropy’ for d(i)) How Science Works Exam Style Questions A2 Chemistry (Nelson Thornes) AQA Chemguide 164 – 173 Born Haber lattice s-cool: Chemical Energetics LUND 7 May 2017 5 A2 Unit 5 Energetics, Redox and Inorganic Chemistry Free Energy and Entropy it seems reasonable that exothermic reactions can occur spontaneously, after all the products are relatively more stable than the reactants (stronger bonds) obviously the rate of the process will depend upon the kinetic barrier (EA) – i.e. kinetic feasibility and they could be slow e.g. iron rusting or extremely slow e.g diamond into graphite spontaneously thus means it will happen under a given set of conditions but not necessarily in an instant and it could be almost infinitely slow a small proportion of spontaneous reactions however are endothermic e.g. solvating NH4NO3(s) at first sight this seems counter intuitive as endothermic reactions require a transfer of heat from their surrounding resulting in products that are relatively less stable (higher stored chemical energy) in terms of enthalpy so why doesn’t NH4NO3(aq) spontaneously undissolve as this would be exothermic and also be kinetically more feasible relative to the forward reaction (lower EA) why does it only go one way (assuming the solubility limit has not been exceeded) clearly there must be another factor in the viability of a chemical (and physical) process that determines whether it will occur spontaneously or not Entropy (S) chemical and physical change are governed by the laws of probability the degree of disorder (ways of distributing energy) within a system is called Entropy the greater the degree of disorder in a system the greater the entropy e.g. state changes solvation diffusion complexity of molecules – increases way to distribute energy hence greater entropy temperature – entropy is increased if it is raised and visa-versa the second law of thermodynamics states that all viable chemical and physical changes result in an increase in the TOTAL entropy in the Universe hence all spontaneous reactions result in an increase in TOTAL entropy irrespective of whether they are exothermic or endothermic an endothermic reaction can thus be spontaneous IF it results in an increased degree of randomness (of distribution of energy) in the universe LUND 7 May 2017 6 A2 Unit 5 Energetics, Redox and Inorganic Chemistry ΔS heat energy flowing into a system will increase its entropy – more available energy levels – more available ways in which energy can be distributed between the particles for an endothermic reaction lowering the temperature reduces entropy but the change from reactants to products could increase entropy (e.g. solvation). IF there is a NET increase in entropy the reaction may be spontaneous like enthalpy, So is specified for 298K and 100 kPa unlike H it is possible to quantify S (units are JK-1mol-1) since it can be extrapolated from a value of zero at 0K Look ! it is possible to determine the entropy change in the system (the chemicals reacting) ΔS(system) = ∑S(products) - ∑S(reactants) note that the overall entropy may only change slightly if all the chemicals are in the same phase if entropy must increase – how can water condense spontaneously given that it achieves a more ordered state i.e. a decrease in entropy? We must also consider the entropy change in the surroundings to complete the picture – remember it is the TOTAL entropy change that concerns us the total entropy change is expressed by: ΔSototal = ΔSosystem + ΔSosurroundings it is very difficult to calculate the entropy change in the surroundings however, it can be determined from the enthalpy transferred from/to the system at a given temperature (since temperature effects the value of entropy) T ΔS o surroundings = -ΔHosystem energy is transferred from the steam (system) to the surface that it condenses on (surroundings) increasing its entropy such that the overall entropy increases the ligand displacement reactions that you will meet in transition metal chemistry can often be attributable to favourable entropy changes where the total number of particles increases LUND 7 May 2017 7 A2 Unit 5 Energetics, Redox and Inorganic Chemistry Gibbs free energy (G) entropy and enthalpy can be combined in a quantity called Gibbs free energy (G) G o is the standard free energy change (kjmol-1) and quantifies the enthalpy and entropy changes at a given temperature for a reaction to be feasible it must be zero or negative i.e such that ΔSototal is positive ΔGo = - TΔSototal some mathematical magic with the previous equations leads us to: ΔGo = ΔHosystem - TΔSosystem kjmol-1 Remember to divide entropy by a 1000 since its in J not kJ !!! G o effectively determines the feasibility of a reaction reaction feasibility is thus determined by the relative magnitude of the enthalpy and entropy changess at a given temperature temperature has a significant effect on reaction feasibility a reaction first becomes feasible when the temperature has been raised to a value where G o = 0 this is particularly relevant to metal extraction where heating may be necessary to make the reaction feasible not just to increase the rate a feasible reaction is not necessarily spontaneous as there may be a high activation energy involved when G o = 0 an equilibrium will exist in a closed system ΔS Outcome - + G always negative Feasible at ALL temperatures + - G always positive NOT feasible at ANY temperature - - IF > T ΔS then reaction is feasible i.e. at LOWER temperatures + + IF T ΔS > then reaction is feasible i.e. at HIGHER temperatures LUND 7 May 2017 8 A2 Unit 5 Energetics, Redox and Inorganic Chemistry Change of State a system is at equilibrium (e.g. ice/water at 0oC) when: G = 0 = T ΔS if heat goes into the system then the entropy term must increase for G to remain zero i.e. it melts to a more disordered state visa-versa is true the entropy change for evaporation is significantly higher than for melting as the increase in randomness is more significant Summary Questions Exam Style Questions A2 Chemistry (Nelson Thornes) AQA Chemguide Page 183 Page 185 Page 246 178 – 183 Entropy LUND 7 May 2017 9 1-3 2–4 1, 2 A2 Unit 5 Energetics, Redox and Inorganic Chemistry Periodicity Elements of Period 3 Structure Electrical Conduction Na Mg giant metallic Al good, delocalised (mobile) eincreases Na Al as more e- delocalised Si Giant Covalent P S Cl Simple Molecular Ar Atoms poor, electrons localised in covalent bonds so not free to move through structure poor Melting Point high, strong metallic bonding, strong forces of attraction throughout entire structure increases Na Al as more e- delocalised higher, strong covalent bonding between atoms low, strong covalent bonds between atoms, but weak intermolecular forces between Molecules very low Mpt/oC 1410 44 -189 98 651 660 113 -101 silicon has a similar structure to diamond – each silicon has 4 covalent bonds silicon is important in the electronics industry sodium is used in streetlights and as a coolant in nuclear reactors magnesium and aluminium are important structural materials often used in alloys chlorine is a product of the salt-alkali industry and has numerous uses argon is used in lighting and lasers sulphur is a raw material for the manufacture of sulphuric acid Reaction of the elements of Period 3 with water Na vigorous reaction – darts about the surface – dissolves to form a very alkaline solution Na(s) + 2H2O(l) 2NaOH(aq) + H2(g) Mg very slow with cold water, but more readily with steam Mg(s) + H2O(l) MgO(s) + H2(g) Cl Unit 2 revision only chlorine reacts with water to produce a 0 -1 +1 mixture of two acids by disproportionation Cl2(g) + H2O(l) HCl(aq) + HOCl(aq) (moist blue litmus is first turned red by chlorine gas then bleached) LUND 7 May 2017 10 A2 Unit 5 Energetics, Redox and Inorganic Chemistry Reactions of the Elements of Period 3 with Oxygen reactivity (generally) decreases across the period argon does not react at all chlorine-oxygen compounds do exist but are not formed by direct combination the metals become coated in an oxide layer – hence appear dull sodium must be stored under oil due to its reactivity aluminium oxide forms a protective layer around aluminium protecting it from further corrosion red phosphorus needs heating to react with oxygen but its allotrope, white phosphorus, is stored under water as when dry it burns spontaneously in oxygen the oxide formed contains the element in its highest oxidation state except for sulphur Na burns very vigorously with a yellow flame Mg burns very vigorously with a white 2Mg(s) + O2(g) 2MgO(s) flame – white MgO ash Al initially vigorous 2Al(s) + 3O2(g) 2Al2O3(s) Si slow Si(s) + O2(g) SiO2(s) P vigorous – white fumes of P4O10 P4(s) + 5O2(g) P4O10(s) phosphorus(V) oxide S melts, blue flame – forms SO2 a S (s) + O2(g) SO2(g) colourless choking acidic gas Summary Questions A2 Chemistry AQA (Nelson Thornes) Chemguide 4Na(s) + O2(g) 2Na2O(s) Page 191 1–3 186 - 188 Period 3 elements LUND 7 May 2017 11 silicon(IV) oxide A2 Unit 5 Energetics, Redox and Inorganic Chemistry Periodicity of Oxides Period 3 Oxides Physical State (298K) Mpt/oC Structure Na2O Mg Al2O3 SiO2 2852 2072 – lower higher – size since and charge of different cation structure Giant Ionic Lattice (Al2O3 has a degree of covalency – small ion + large charge) 1275 Basic Water 1610 Giant Covalent similar to diamond Amphoteric Simple Molecular Acidic Acid Alkali Na2O + H2O 2NaOH oxide ion is hydrolysed very soluble pH 14 acid + metal oxide salt + MgO + H2O Mg(OH)2 a weakly alkaline solution water only slightly soluble pH 9 Al2O3 + 3H2O + 2OH 2[Al(OH)4]SiO2 + 2OH- SiO32(silicate ion) + H2O P4O10 + 12OH- 4PO43(phosphate(V) ion) + 6H2O SO2 + 2OH- SO32(sulphite ion) + H2O SO3 + 2OH- SO42(sulphate ion) + H2O Si S 580 ? SO3 (SO2) liquid (gas) 17 (-73) inc. IMF Al P P4O10 Solid Nature of Oxide Reaction of Oxide Na MgO P4O10 + 6H2O 4H3PO4 phosphoric(V) acid pH 0 SO2 + H2O H2SO3 sulphuric(IV) acid pH 3 SO3 + H2O H2SO4 sulphuric(VI) acid pH 0 aluminium oxide acts as an abrasive (as corundum) due to the strong bonding aluminium/magnesium oxides are used as a refractory material (furnace) melting point silicon(IV) oxide > carbon dioxide (strong covalent bonding throughout the structure rather than simple molecular lattice held together by weak intermolecular forces) you should also be able to write a 3 step equation for phosphoric acid and water Summary Questions Exam Style Questions A2 Chemistry AQA (Nelson Thornes) Chemguide Page 193 Page 194 Page 249 1–3 1-6 7 189 - 193 Period 3 oxides LUND 7 May 2017 12 A2 Unit 5 Energetics, Redox and Inorganic Chemistry Oxidation Numbers oxidation is the LOSS of electrons, reduction is the gain of electrons OILRIG oxidising agents (oxidants) accept electrons and are themselves reduced reducing agents (reductants) donate electrons and are themselves oxidised Oxidation State this is a ‘book keeping’ method of the effective control of electrons used in bonding elements = 0 oxidation state of elements in simple ions = charge on ion oxidation state of elements in polyatomic ions = charge on ion oxidation state of elements of a compound = 0 the relatively more electronegative element is assigned the negative oxidation state hydrogen = +1 (except in metal hydrides where it is -1) oxygen = -2 (except in peroxide O22-) where it = -1) group 1 metals = +1 group 2 metals = +2 fluorine = -1 (even with oxygen, which is +2 in OF2) Aluminium = +3 metals are always positive in a compound or polyatomic ion maximum possible oxidation state = group number (note not always possible for various reasons – see later) oxidation numbers and nomenclature e.g. cobalt(II) nitrate(V), phosphorus(V) oxide take care not to mix up charge and oxidation numbers in polyatomic species e.g. a sulphate(IV) ion does NOT have a 4- charge (the IV refers to the oxidation state of the sulphur) changes in oxidation numbers can be used to identify redox reactions in inorganic and organic reactions e.g. metal or halogen displacement reactions we specifically refer to an element in a species (e.g. ‘the iron in Fe2O3 is reduced’) assume that multiple instances of an element in a species have the same value (e.g. both carbons in ethene are -2) OXIDATION REDUCTION oxidation number becomes relatively more positive oxidation number becomes relatively more negative A2 Chemistry AQA (Nelson Thornes) Chemguide 196 - 197 oxidation number LUND 7 May 2017 13 A2 Unit 5 Energetics, Redox and Inorganic Chemistry Balancing Redox Equations (Acidic conditions) protocol for constructing half equations 1 2 3 4 5 try doing these important half equations (either way round): (i) (ii) (iii) (iv) (v) I have written some answers. Try to find them and do the bonus STAR question get the species correct (and number e.g. 2Cr3+) balance the oxygen by adding H2O(l)’s to the side with least O’s balance the hydrogen’s using H+(aq)’s to the side with least H’s balance the charge on each side by adding e- to relatively more positive side add state symbols Fe3+(aq)/ Fe2+(aq) MnO4-(aq)/Mn2+(aq) Cr2O72-(aq)/Cr3+(aq) S4O62- (aq)/ S2O32-(aq) (tetrathionate/thiosulphate) CO2(g)/C2O42-(aq) (ethanedioate ion) HSW: This titration can be used to investigate the % of copper in a coin – your teacher will explain how if you ask now try combining these half equations: (i) (ii) (iii) (iv) (v) iodine and thiosulphate ions iron (II) ions and manganate(VII) ions iron(II) ions and dichromate(VI) ions manganate(VII) ions and ethanedioate ions dichromate(VI) ions and ethanal Summary Questions Exam Style Questions HSW: This titration can be used to standardise solutions of potassium dichromate(VI) or potassium manganate(VIII) 1 – 2 (Q2 should have equilibria arrows) Page 199 Page A2 Chemistry AQA (Nelson Thornes) Chemguide HSW: These titrations can be used to investigate the % of iron in an iron tablet or iron in tea (after some preparatory steps) 197 - 199 redox equation LUND 7 May 2017 14 A2 Unit 5 Energetics, Redox and Inorganic Chemistry Redox equations in alkaline conditions this is included to allow you to work out rather than rote learn redox reactions for some organic (e.g. Fehling’s and Tollen’s), transition metal reactions (e.g. chromium) in alkaline conditions and certain reactions associated with batteries writing half equations for redox reactions under alkaline conditions is a little more tricky than under acidic conditions – but here is a useful ‘cheat’ do it exactly as if it was in acidic conditions then cancel out the hydrogen ions by adding hydroxide ions equally to both sides you must know and use the formula of the species containing the atom(s) to be oxidised/reduced as it exists in alkaline conditions lets consider Fehling’s (see Module 4) in which copper(II) ions are reduced to copper(I) which exists as copper(I) oxide and an aldehyde is oxidised to a carboxylic acid (why would you not smell this?) using the correct species for alkaline conditions but as if in an acidic solution you should arrive at: RCHO(aq) + 2Cu2+(aq) + 2H2O(l) RCOOH(aq) + Cu2O(s) + 4H+(aq) now the modification add OH-(aq) to BOTH sides in order to cancel 2H+ subsequently cancel out H2O’s to arrive at: RCHO(aq) + 2Cu2+(aq) + 4OH-(aq) RCOOH(aq) + Cu2O(s) + 2H2O(l) this method also works perfectly well if applied to half equations try using it to derive the equations met in Periodicity of oxides reacting with alkali from the known reactant and product LUND 7 May 2017 15 A2 Unit 5 Energetics, Redox and Inorganic Chemistry Electrode potentials and the Electrochemical Series dynamic equilibrium exists between a metal and its ions in solution which results in a potential on the metal Mn+(aq) + ne- M(s) this potential depends upon the position of the equilibrium and cannot be measured absolutely it will be different for a more or less reactive metal so there will be a potential difference, which is measurable, between the two pieces of metal (try chewing a piece of aluminium if you have a metal filling – you’ll get the idea) the relatively more reactive a metal, the relatively more biased the above equilibria will be to the LHS, the relatively more negative the potential on that metal will be you should know the practical arrangement of a single cell made up of two half cells (see figure 3 on page 200) Zn2+(aq)|Zn(s) and Cu2+(aq)|Cu(s) Note: the relatively more oxidised form is shown on the LHS by convention (in practice both reactions can go either way depending upon what they are combined with) the pieces of metal are called electrodes (in this case the cathode is the +ve terminal unlike in electrolysis where it’s the –ve terminal since the cathode is defined as the electrode where reduction takes place) the wire allows electron flow between the two electrodes the salt bridge provides mobile ions to complete the circuit potassium nitrate (or similar) used in the salt bridge as the ions are unlikely to partake in electron exchange or precipitation oxidation takes place at the negative electrode (zinc in this case) reduction takes place at the positive electrode (copper in this case) given that the copper is the +ve electrode (by + 1.10V in theory) electrons flow to it so the reactions that are taking place in the two half cells are: Cu2+(aq) + 2e- → Cu(s) Zn(s) → Zn2+(aq) + 2e- and in effect the more reactive metal (better reducing agent) has displaced the less reactive metal from its solution A2 Chemistry AQA (Nelson Thornes) Chemguide 200 - 201 redox equilibria, half cells LUND 7 May 2017 16 A2 Unit 5 Energetics, Redox and Inorganic Chemistry Standard Hydrogen Electrode there are a huge number of different half cells that can be constructed (some of which don’t involve a metal in the redox reaction thus requiring an inert platinum electrode) to have comparisons between them all would just be silly so we use an electrode potential equivalent of sea level to which all are then compared this works exactly like in geography where relative heights of mountains and depths of ocean trenches are given allowing differences to be calculated similarly, the standard electrode (reduction) potential Eo for each half cell is measured relative to the standard hydrogen electrode (SHE), which is defined as 0V under standard conditions: 25oC 100Kpa 1.0 moldm-3 H+(aq) CARE – must be strong acid, and 0.5 moldm-3 if H2SO4 a diagram of the standard hydrogen electrode (PRIMARY STANDARD) and how it is used is shown in figure 6 on page 201 which you must be able to draw a platinum electrode is used as this will not undergo a redox reaction thus will not alter the potential of the hydrogen electrode it is covered with finely divided platinum (termed ‘platinized platinum’) to increase surface area and increase rate all solutions will be 1.0 moldm-3 and 25oC as other values will alter the relative position of the equilibria and therefore the absolute and hence measured potential difference in practice the Calomel electrode A SECONDARY STANDARD is used for convenience and is initially calibrated against the SHE at a value of +0.27V Hg2Cl2(aq) + - 2e 2Hg(l) + - 2Cl (aq) the voltmeter used has a high resistance hence a low current is drawn so a true reading of the potential difference is made if a large current is drawn the concentrations of the ions in solution will change thus upsetting the equilibria and consequentially changing the value being measured A2 Chemistry AQA (Nelson Thornes) Chemguide 200 - 1 hydrogen electrode, non-metal systems, calomel LUND 7 May 2017 17 A2 Unit 5 Energetics, Redox and Inorganic Chemistry Electrochemical Series the electrochemical (reactivity) series is related to standard electrode potentials. standard electrode potentials can be used to predict the relative feasibility of a reaction by convention the standard electrode potentials are always quoted for the reduction process the more +ve its value the relatively more likely the forward reaction is going to occur i.e. the stronger the oxidant (on the left) the more -ve its value the relatively more likely the reverse reaction is going to occur i.e. the stronger the reductant (on the right) It thus follows that… any species on the LHS can potentially oxidise any species on the RHS with a relatively more negative electrode potential any species on the RHS can potentially reduce any species on the LHS with a relatively more positive electrode potential thus relatively more reactive metals will have a relatively more –ve electrode potential lets use a familiar idea to reinforce this – ‘a more reactive metal can displace a less reactive metal’ e.g. iron nails in copper(II) sulphate solution (not the other way around) Cu2+(aq) + 2e- Cu(s) Eo = + 0.34V Fe2+(aq) + 2e- Fe(s) Eo = - 0.44V which of the above metals will dissolve in 0.5M sulphuric acid? NOTES: 1. 2 the number of electrons lost or gained is not a factor in their relative availability other factors, e.g. kinetics, may prevent a feasible reaction from occurring i.e electrode potentials do not indicate the rate of a reaction, just its feasibility (for example where a solid is involved, or where two ions of the same charge are the reactants) LUND 7 May 2017 18 A2 Unit 5 Energetics, Redox and Inorganic Chemistry Representing Cells convention for representing cells (with the relatively +ve half cell on the RHS): R O O R eZn(s)| Zn2+(aq)||Cu2+(aq)|Cu(s) single line is boundary between phases commas are used between same phase (e.g. Fe3+(aq),Fe2+(aq)) double line = salt bridge (sometimes shown as a single dashed lines in some sources) the emf quoted is the right hand side relative to the left hand side Eocell = EoRHS the hydrogen half cell can be represented in two different ways life is easier if you put it on the side that provides a positive value for the cell overall as it is written Pt(s)| H2(g)|H+(aq)||other half cell other half cell||H+(aq)|H2(g)|Pt(s) e.g. for Zn2+(aq)|Zn(s) the electrons flow from the zinc half cell to the hydrogen half cell (Zn is higher in the reactivity series than hydrogen): Zn(s)| Zn2+(aq)||H+(aq)|H2(g)|Pt(s) Eocell = +0.76V - EoLHS zinc is –0.76V i.e its – ( - 0.76) EoRHS - EoLHS 0.00V - Zn half cell the standard electrode potentials of ions of the same element in different oxidation states can be measured e.g. Eo Fe3+/Fe2+ = + 0.77V 1 2. 3. What will be used as the electrode? Show the cell diagram What concentrations of iron sulphate solutions will you mix together? Summary Questions Exam Style Questions 1–2 4 Page 203 Page 213 A2 Chemistry AQA (Nelson Thornes) Chemguide 203 Electrochemical LUND 7 May 2017 19 A2 Unit 5 Energetics, Redox and Inorganic Chemistry Electrode Potentials and Reaction Direction the feasibility of a reaction requires electrons to flow in the right direction – the reverse reaction is NOT feasible under standard conditions Reducing agent provides e- and is itself oxidised R O O R Oxidising agent accepts eand is itself reduced ethe oxidising agent must have a relatively more positive electrode potential the reducing agent must have a relatively more negative electrode potential remember that the electrode potentials are quoted for unimolar concentrations under standard conditions and will be modified through changes in: concentration pH temperature the effect of changes can be determined by applying LCP to the feasibility of a reaction e.g. increasing the concentration of one of the reactants involved in the oxidising half will increase its electrode potential thus feasibility i.e. the forward reaction is more favoured similarly increasing the concentration of one of the reactants involved in the reducing half will decrease its electrode potential thus the reverse reaction is favoured our technician wishes to prepare chlorine gas based on the electrode potentials given below and you need to suggest how this can be achieved Cl2 + 2e- 2Cl- Eo = +1.36 V MnO2 + 4H+ + 2e- Mn2+ + 2H2O Eo = +1.23 V when you study the variable oxidation states of chromium later on you will note that alkaline conditions are used during oxidation – once again this modifies the associated electrode potentials and hence reaction feasibility consider the feasibility of coating a copper/nickel coin with zinc as a first step to making a ‘gold’ coin based on standard electrode potentials during the operation of a battery the conditions change such that it goes ‘flat’ Summary Questions Exam Style Questions 1–4 1–3 3 Page 207 Page 212 Page 246 A2 Chemistry AQA (Nelson Thornes) Chemguide 204 - 7 Electrochemical, vanadium, reaction feasibility LUND 7 May 2017 20 A2 Unit 5 Energetics, Redox and Inorganic Chemistry Learn the Daniel cell, zinc-carbon battery, lead acid battery and hydrogen fuel cell. Develop an understanding of the rest. Electrochemical Cells 1800 – Alessandro Volta invents a primitive battery called a voltaic pile consisting of alternative layers of silver (or copper) and zinc discs separated by cardboard soaked in salt water Non-rechargeable Cells (Primary Cells) The electrochemical reaction is not reversible (cells can be used only once) as when discharging the cell the chemicals are permanently changed. Daniell Cell a ‘wet cell’ invented by John Daniell in 1836 the porous pot was used (in place of the salt bridge shown below) to allow the ions to migrate when the battery was operating whilst preventing the solutions from mixing or without this barrier, even when no current was drawn, the ZnSO4 copper ions would be reduced at the zinc anode thus shortening the battery's life negative ions will migrate in the same direction as the electrons around the circuit and positive ions visa-versa it was widely used at telegraph stations in the 19th century, however, its portability was limited due to the liquid electrolytes that it contained THE CATHODE THE ANODE Reduction takes place as the positive pole of the battery (half-cell with the highest electrode potential) accepts electrons from the external circuit Copper metal deposits on the cathode – hence its mass increases Oxidation takes place as the negative pole of the battery (half-cell with the lowest electrode potential) releases electrons to the external circuit Zinc goes into aqueous solution (hence the zinc plate loses mass) The solution becomes more dilute hence the blue colour fades eYou could write lots of notes but ideally you should be able to work it all out from this schematic LUND 7 May 2017 Zn(s)| Zn2+(aq)||Cu2+(aq)|Cu(s) Eocell = EoRHS 0.34 - EoLHS - 0.76 Note that, unlike in electrolysis, the cathode is the positive terminal in a battery. This is because the cathode is specified as the electrode at which reduction takes place. = +1.10V Forgetting the double minus is a common source of error 21 A2 Unit 5 Energetics, Redox and Inorganic Chemistry STANDARD Zinc – Carbon ‘Dry Cell’ 1887 - Carl Gassner patented a dry cell variant of the 1866 Leclanché wet cell the electrodes are zinc and carbon, with an acidic paste between them zinc serves as both the anode and the container, allowing the battery to be completely self-contained and in effect more portable and practical than wet cells it could be used in any position as well rather than on a flat surface and the risk of leaking was greatly reduced the cathode is a mixture of powdered (surface area contact) manganese dioxide and graphite surrounding a solid graphite rod the electrolyte is a paste of ammonium chloride inside the zinc can + CATHODE: Eo = +0.7V - ANODE: Eo = -0.8V 2NH4+(aq) + 2e- → H2(g) + 2NH3(aq) Zn(s) → Zn2+(aq) + 2e- This means the casing gets thinner in use 2NH3(aq) + Zn2+(aq) → [Zn(NH3)2]2+(aq) prevents ammonia leaking (what shape is the complex ion) +4 +3 2MnO2(s) + H2(g)→ Mn2O3(s) + H2O(l) prevents a pressure build up from H2(g) overall reaction in a STANDARD zinc-carbon cell is: Note that overall it’s the manganese that is reduced Zn(s) + 2MnO2(s) + 2NH4+(aq) → Mn2O3(s) + Zn(NH3)22+(aq) + H2O(l) whilst zinc-carbon batteries are inexpensive they have very low power density so are only useful in devices that draw very little current the zinc container also becomes thinner when used as the zinc is oxidised it also thins when not used as ammonium NH4+(aq) + H2O(l) ⇋ H3O+(aq) + chloride is acidic and slowly reacts with the zinc NH3(aq) which may lead to leakage hence the service/shelf life of the battery is relatively short the terminals of the battery are made of tin plated steel or brass to prevent the exposure the zinc, not allowing it to corrode as quickly, thus adding to the total battery life the seal usually is made of asphalt pitch, wax, or plastic to allow the cathode mix (when the battery gets warm) to expand without rupturing the casing LUND 7 May 2017 22 A2 Unit 5 Energetics, Redox and Inorganic Chemistry ‘HEAVY DUTY’ Zinc Chloride Cell zinc chloride cells are an improvement on the original zinccarbon cell giving a longer life (about 50% due to a variation in the chemical mix and greater mass) electrolyte ZnCl2 paste (cf NH4Cl in standard Zn-C) - ANODE: + CATHODE: +4 +3 2MnO2(s) + H2O(l) + 2e- → Mn2O3(s) + 2OH-(aq) Zn(s) → Zn2+(aq) + 2eoverall reaction in a HEAVY DUTY zinc-chloride cell is: Zn(s) + 2MnO2(s) + H2O(l) → Mn2O3(s) + Zn2+(aq) + 2OH-(aq) these were originally marketed around 50 years ago as "Heavy Duty” batteries, but since this term is not standardised it is a misleading as they are much inferior to alkaline batteries which have since been introduced and are around 300% better Alkaline Cell Battery sold under brand names such as ‘Duracell’ ‘Energizer’ these are more expensive than, but last considerably longer than, ‘ordinary zinc-carbon cells’ the cathode is manganese(IV) oxide powder the anode is zinc powder (more surface area for increased rate of reaction therefore increased electron flow to allow for heavy duty usage the electrolyte is potassium hydroxide paste (hence ‘alkaline’) + CATHODE: - ANODE: +4 +3 2MnO2(s) + H2O(l) + 2e → Mn2O3(s) + 2OH-(aq) Zn(s) + 2OH- (aq) → ZnO(s) + H2O(l) + 2e- Regenerated overall reaction is: Zn(s) + 2MnO2(s) → Mn2O3(s) + ZnO(s) no gases (which insulate the electrodes) are produced which is one reason why they don’t suffer from a voltage drop as do zinc-carbon batteries when worked hard NOTE: ‘Button’ batteries consist of a range of different types of system e.g. silver oxide, alkaline and are a structural form, for a specific use (compact size/long life), rather than a particular chemistry so are not described in this guide. At one time mercuric oxide was used but this has ceased for obvious reasons. LUND 7 May 2017 23 A2 Unit 5 Energetics, Redox and Inorganic Chemistry Rechargeable Cells (Secondary Cells) The electrochemical reaction is not reversible (cells can be used only once) as when discharging the cell the chemicals are permanently changed Lead-acid battery 1859 - Gaston Planté - the lead-acid cell: the first rechargeable battery the cathode is a lead-antimony alloy grid coated with lead dioxide the anode is a lead-antimony alloy grid coated in spongy lead (its porous to increase surface area) the electrolyte is ~ 6M sulfuric acid in which the plates are immersed at the anode lead combines with sulphate ions to create lead sulfate and release electrons as the battery discharges, both plates build up lead sulphate and water builds up in the acid thus diluting it the voltage is about 2 volts per cell, so by combining six cells in series you get a 12-volt battery upon discharging the following reactions take place: + CATHODE: Eo = +1.68V PbO2(s) + 4H+(aq) + SO42-(aq) + 2e- → PbSO4(s) + 2H2O(l) - ANODE: Eo = -0.36V Pb (s) + SO42-(aq) → PbSO4(s) + + 2eOverall: Eo = 2.04V PbO2(s) + 4H+(aq) + 2SO42-(aq) + Pb(s) → 2PbSO4(s) + 2H2O(l) upon charging the reactions are reversed and this takes place when the car is running (using the alternator) so that lead sulphate does not build up hence giving a ‘flat battery’ even when not in use, leakage of current takes place so there is a net usage of sulphuric acid hence the need for it to be checked and topped up when the vehicle is serviced given the emphasis on fuel economy the battery must not add too much weight to the vehicle but must provide enough power to start the car even in cold weather for most of the last century these batteries have become standard for starting cars as they can produce short bursts of a high current for many decades and are easy and cheap to manufacture however they cannot be used to power a vehicle for longer journeys as they provide relatively little energy per kilogram and suffer power loss as insulating lead sulphate builds up why do you think a car battery could explode if it is overcharged LUND 7 May 2017 24 A2 Unit 5 Energetics, Redox and Inorganic Chemistry Nickel-Cadmium Cells ‘Nicads’ 1950s - Nickel-Cadmium (NiCd) first appeared these are much more portable than lead-acid batteries but are more expensive they are manufactured in sizes/voltages to act as direct replacements for cheaper zinc-carbon batteries the fact that they can be recharged make them economical in the longer term the cathode is made from nickel(III) oxyhydroxide the anode is made from cadmium the electrolyte is potassium hydroxide when discharging the reactions are (reversed when charging): + CATHODE: Eo = +0.52V NiO(OH)(s) + H2O(l) + e- → Ni(OH)2 (s) + OH-(aq) Note some sources state Ni(OH)3 here and omit H2O’s to balance Overall: - ANODE: Eo = -0.81V Cd(s) + 2OH-(aq) → Cd(OH)2(s) + 2e- Eo = 1.33V 2NiO(OH)(s) + 2H2O(l) + Cd(s) → 2Ni(OH)2 (s) + Cd(OH)2(s) cadmium is toxic so there are environmental issues regarding disposal Nickel Metal Hydride Cells Comparison of the discharge 1986 - NiMH battery was patented as a voltage of an alkaline battery (red) bi-product from research on the and a NiMH battery (blue). The green line is the voltage at which storage of hydrogen for use as an the battery is considered dead alternative energy source in the 1970s some metallic alloys were observed to form hydrides that could capture (and release) Hydrogen in volumes up to nearly a thousand times their own volume compared to lead-acid and NiCd, NiMH batteries have a higher storage capacity they are more expensive than lead-acid and NiCd, but they are considered better for the environment (lead and cadmium are toxic) – prices are falling but lithium ion batteries are starting to gain some of the market however, a NiCd battery has a lower self-discharge rate i.e. they hold their charge better as with NiCd’s they offer slightly under 1.5V so may not work with some devices designed to operate off the 1.5V of alkaline or zinc-carbon batteries LUND 7 May 2017 25 A2 Unit 5 Energetics, Redox and Inorganic Chemistry the cathode is made from nickel(III) oxyhydroxide (the same as NiCd’s) the anode is made from an alloy of rare earth metals that can soak up hydrogen atoms with the general formula AB5 where A can be lanthanum and B nickel the electrolyte is potassium hydroxide + CATHODE: Eo = +0.52V NiO(OH)(s) + H2O(l) + e- → Ni(OH)2 (s) + OH-(aq) Eo = -0.83V - ANODE: AB5H(s) + OH-(aq) → AB5(s) + H2O(l) + eOverall: Eo = 1.35V NiO(OH)(s) + AB5H(s) → Ni(OH)2 (s) + AB5 Lithium ion Cells compared with NiMH and NiCd batteries of the same sizes or weights, Lithium Ion batteries are designed to deliver the highest energy output a single cell voltage is 3.7V, 3 times that of NiMH batteries so a simpler battery configuration and better space utilization is achievable in devices such as cameras they are relatively expensive as a computer chip is required to control charging and discharging but do offer a high capacity (hence reducing mass/size) Li Ion batteries contain no toxic heavy metals, such as mercury, cadmium or lead the cathode is made from lithium cobalt oxide powder the anode is lithium/graphite formulation (a lot of technological development was required to prevent lithium oxidising and costing the electrode with insulating lithium oxide 1996, the lithium ion polymer battery was developed from the lithium ion battery these batteries hold their electrolyte in a solid polymer composite which can’t leak the electrodes and separators are laminated to each other with the whole devices encased in a flexible wrapping instead of a rigid metal casing, which means such batteries can be specifically shaped to fit a particular device Fuel Cells The Hydrogen Economy fossil fuels are at present the most economical way to power transportation however, price rises commensurate with supply and demand, plus pollution issues such as the greenhouse gas CO2 and acidic nitrous oxides (from atmospheric N2 + O2 in a hot engine) etc are driving the need for an alternative LUND 7 May 2017 26 A2 Unit 5 Energetics, Redox and Inorganic Chemistry an alternative fuel is hydrogen which if combusted does not give the pollution problems associated with hydrocarbons, the product is water hydrogen can be obtained from the electrolysis of sea water in the longer term but at present most hydrogen is still obtained from fossil fuels by steam methane reforming this reacts steam with methane (natural gas) over a heated nickel catalyst to produce hydrogen and carbon monoxide energy is obviously required to obtain the hydrogen so fuel cells are not the ‘free’ energy from water that is often suggested however, given that nuclear reactors can’t be turned off, off peak generation could be one of the means of generating less expensive hydrogen along with wind, hydro, solar and tidal whichever, there may still be some pollution associated with hydrogen production Hydrogen Fuel Cell in a battery the chemical energy is stored within the electrodes and the solution in a fuel cell hydrogen (fuel) and air (oxygen) are fed into the cell in a similar way that petrol and air are fed into an internal combustion engine the difference is that the chemicals are not combusted but react to produce electricity directly this is more efficient than combusting the hydrogen (chemical → heat → kinetic → electrical energy) since a continuous supply of hydrogen is provided the voltage output remains constant a typical fuel cell consists of two platinum electrodes these also act as catalysts to assist the decomposition of hydrogen molecules the electrodes are separated by a polymer electrolyte (proton exchange membrane) through which hydrogen ions can migrate whilst the gases are kept apart + CATHODE: Eo = +1.2V - ANODE: Eo = 0.0V 4H+(aq) + O2(g) + 4e- → 2H2O(l) H2(g) → 2H+(aq) + 2eZn(s) → Zn2+(aq) + 2e- Overall: Eo = 1.2V O2(g) + 2 H2(g) → 2H2O(l) a major issue is the storage and transportation of liquid hydrogen research is currently being undertaken to develop hydrocarbon fuel cells so that car manufacturers can rely of normal fuel tanks this is more complex as it requires preliminary reforming of the fuel within the vehicle many other alternatives are currently under investigation it is likely in the interim that fuel cell/battery/petrol hybrids will be employed to a greater extent LUND 7 May 2017 27 A2 Unit 5 Energetics, Redox and Inorganic Chemistry Overview batteries reduce the need for expensive cabling and can provide power supplies to remote places non-rechargeable batteries are cheap and can be manufactured in all sizes e.g. button batteries for watches, larger batteries for torches etc however, they are thrown away afterwards along with energy and resources used in their manufacture rechargeable batteries are more expensive initially but less resources are wasted in the longer term they are vital in solar powered devices (and similar devices intent on storing power for future use) the lead and nicad’s contain toxic chemicals thus there are disposal issues – they must not go to landfill sites and must be recycled neither are suitable for vehicles as they add too much mass and alternatives are being investigated NiMH and lithium ion batteries are more environmentally friendly as they do not contain toxic heavy metals sodium-sulphur batteries do offer a better power per kg output but have to operate at 300oC another possibility is metal-air batteries (possible metals include aluminium and zinc) a major issue is power density i.e. how much energy can be stored per kilogram of battery, particularly where small size (e.g. MP3 players) or small mass (transport) is required most batteries have performance that varies with temperature (either way) fuel cells only produce water (spacecraft can use fuel cells to provide drinking water) and will eventually become the standard power source for vehicles which will reduce CO2 emissions IF the hydrogen can be produced cheaply without fossil fuels they provide a more efficient means of converting chemical energy into electrical energy since it is direct rather than by a turbine there are however problems associated with the transportation and storage of hydrogen and the means of refuelling the vehicle you might be asked to calculate a voltage from electrode potentials but should be aware that the actual value will be unlikely to be this as it will not be operating under standard conditions e.g variation in temperature Exam Style Questions Page 246 A2 Chemistry AQA (Nelson Thornes) 3 200 – 201, 208 - 211 tbd LUND 7 May 2017 28 A2 Unit 5 Energetics, Redox and Inorganic Chemistry Transition Metals transition metal - d block elements that are able to form ions with a partially filled d sub shell Sc is not a transition metal because its ion, Sc3+, is iso-electronic with Ar i.e. no d electrons Zn is not a transition metal because its ion, Zn2+, has full d sub-shell the electronic configuration for atoms and ions (remember to write 3d then 4s !) are written left to right in order of increasing energy whilst the 4s subshell is initially of a lower energy than an unoccupied 3d, hence filled first, adding electrons to the 3d pushes the 4s electrons away from the nucleus thus raising their energy don’t forget that copper and chromium are not systematic note that 4s electrons are always lost first when ions are formed and so first series transition metal ions never have any 4s electrons present – so don’t even show 4s (4s0 is incorrect) if you are showing ions using the electrons in boxes nomenclature then note that paired d electrons are lost first (check Fe3+ and Co2+ for example) as mutual repulsion makes these easier to remove Physical Properties typical metals i.e. malleable; ductile; good electrical and thermal conductors; all explained by the same ideas taught in Foundation Chemistry – remind yourself of these. melting point is higher than s block metals since there are more delocalised electrons holding structure together – which also explains their greater mechanical strength Chemical Properties variable oxidation states coloured ions (coloured rocks e.g. hæmatite, malachite, typically include transition metal compounds) catalysts complex ions Summary Questions Page 215 A2 Chemistry AQA (Nelson Thornes) Chemguide Q 1, 2 214 - 5 Transition LUND 7 May 2017 29 A2 Unit 5 Energetics, Redox and Inorganic Chemistry Complex Ions a complex ion is a metal ion surrounded by ligands ligands are molecules or ions which form dative (co-ordinate bonds) by donating electron pair(s) - lone pair donor - to a central metal ion – lone pair acceptor the coordination number is the number of coordinate bonds formed with the central metal ion NOT necessarily the number of ligands ligands (electron pair donors) are Lewis bases transition metal ions (electron pair acceptors) are Lewis acids Unidentate ligands form only one co-ordinate bond with the TM ion Neutral NH3 Negative OHSCNPr- ammine H2O aqua Cl- hydroxo thiocyanato chloro CN- cyano pavarotto (a rather large ligand not on the syllabus) alphabetical order of name (prefixes i.e. di, tri etc ignored) oxidation state of transition metal is given by roman numerals and this will only be the same as the charge if all the ligands are neutral name of metal is in Latin if complex ion is –ve copper = cuprate lead iron = ferrate vanadium manganese = manganate chromium zinc = zincate aluminium e.g. = = = = plumbate vanadate chromate aluminate tetrachlorocuprate(II) ion [CuCl4]2- (ends in ‘ate’ because it is anionic) hexaaquacopper(II) ion [Cu(H2O)6]2+ NOT 3+ tetraamminedicyanocobalt(III) ion number of each [Co(NH3)4(CN)2]+ ligand central metal ion and its oxidation type of ligands LUND 7 May 2017 30 number A2 Unit 5 Energetics, Redox and Inorganic Chemistry Bi-dentate ligands two coordinate bonds per ligand Neutral 1,2-diaminoethane (‘en’) benzene-1,2-diol Negative ethandioate (oxalate) ions (C2O42-) benzene-1,2-dicarboxylate two co-ordinate bonds per molecule leading to chelated (crab) complexes this is also called chelation the possibility of a bidentate ligand acting as a bridge between two separate metal ions exists the replacement of unidentate with bidentate ligands is favoured by entropy since the total number of particles increases (see the section on entropy) Multidentate ligands more than two coordinate bonds per ligand e.g. ethylenediamminetetraacetate ion - EDTA4- EDTA complexes are very stable – in effect a protective cage is formed around the transition metal ion thus isolating it from a biological system the replacement of unidentate with multidentate ligands is favoured by entropy since the total number of particles increases (see the section on entropy) Uses of EDTA 1. 2. 3. 4. Summary Questions antidote to Hg/Pb poisoning (traps metal ions) Ca2+ trap in blood transfusions – prevents clotting removal of Ca2+ from hard water (e.g. in shampoo) titimetric determination of metal ion concentration Page 219 A2 Chemistry AQA (Nelson Thornes) Chemguide 2 216 - 8 complex LUND 7 May 2017 31 A2 Unit 5 Energetics, Redox and Inorganic Chemistry Shapes of Complex Ions octahedral whilst the coordination number is 6 there are 8 faces which determines the allocated name based on an octahedral unit tetrahedral e.g. with H2O, NH3, EDTA, en e.g. with Cl- note that only 4 chloro ligands can fit around the central metal ion due to their relatively large size hence the complex ion adopts a tetrahedral geometry linear (often in silver(1) and copper(I) complexes e.g. [Ag(NH3)2]+ Tollens’ reagent [Cu(NH3)2]+ square planar e.g. cis-platin [Pt(NH3)2Cl2] (more on this later) Extra info: Summary Questions Exam Style Questions geometrical and optical isomerism are possible cis [Co(NH3)4Cl2] +(aq) is violet trans [Co(NH3)4Cl2] +(aq) is green Page 219 Page 233 A2 Chemistry AQA (Nelson Thornes) Chemguide 1 3 217 - 8 Complex shape LUND 7 May 2017 32 A2 Unit 5 Energetics, Redox and Inorganic Chemistry Colours of Complex Ions light absorbed depends on the E of electronic transitions to the next vacant energy level E = hh is Planck’s constant however, E(3d4s) in Ti3+ cannot account for lilac colour as E is too high (i.e. uv frequency) in an isolated transition metal ion the d orbitals all have the same energy i.e. they are degenerate however, ligands split the 3d energy level so that E is of a lower value corresponding to the energy of visible light the reason for this is that the electrons donated by the ligand change the electronic environment to different extents for different d-orbitals in different geometrical positions i.e the are all raised in energy but to differing degrees white light incident upon a transition metal solution or solid will have certain wavelengths absorbed in accordance with the value of E when exciting an electron thus removing this colour from the spectrum colour observed is the complementary colour of light absorbed hexaaquacopper(II) ions are blue as red is absorbed (see colour wheel) LUND 7 May 2017 33 A2 Unit 5 Energetics, Redox and Inorganic Chemistry Sc3+ and Zn2+ are not coloured as there are no partially filled 3d sub-shells which is necessary for this to work i.e. to allow the promotion of d-electrons between d-ortbitals colour depends on: central metal ion this is a major factor - obviously oxidation state e.g. Fe(OH)2(s) Fe(OH)3(s) Cr2O72-(aq) [Cr(H2O)6]3+(aq) MnO4-(aq) Mn2+(aq) [Co(NH3)6]2+(aq) [Co(NH3)6]3+(aq) co-ordination number has a significant effect on d-d splitting hence colour change varies size of E and the type of d-d splitting e.g. [Co(H2O)6]2+(aq) [Co(Cl)4]2-(aq) [Cu(H2O)6]2+(aq) [Cu(Cl)4]2-(aq) octahedral geometry yields 2 higher 3 lower tetrahedral geometry yields 3 higher 2 lower type of ligand stronger bonding causes greater d-d spitting hence shorter wavelength absorbed (spectrochemical series (Cl- < H2O < NH3 < en < CN-) e.g. [Cu(H2O)6]2+(aq) [Cu(NH3)4(H2O)2]2+(aq) [Co(H2O)6]2+(aq) [Co(NH3)6]2+(aq) YOU MUST LEARN THESE COLOURS [Cu(H2O)6]2+(aq) Blue [CuCl4]2-(aq) Yellow 2+ Adding cHCl to [Cu(H2O)6] (aq) gives green! [Cu(NH3)4(H2O)2]2+(aq) Deep Blue NH3(aq) definitive test for Cu2+(aq) [Co(H2O)6]2+(aq) Pink [CoCl4]2-(aq) Blue cobalt chloride paper is a test for water [Co(NH3)6]2+(aq) Yellow [Co(NH3)6]3+(aq) [Fe(SCN)(H2O)5]2+(aq) sensitive test for the presence of Fe3+(aq) LUND 7 May 2017 34 Brown Blood Red A2 Unit 5 Energetics, Redox and Inorganic Chemistry Colorimetry you should re-familiarise yourself with the nature of light and why we see colour two ways in which chemicals can interact with light result in: absorption spectra (star light through a a planetary atmosphere, chlorophyll) emission spectra (e.g. flame tests, street lights) absorption in the visible light varies according to the complex ion present, path length and concentration absorption of aqua complexes is relatively weak so colours are not very intense certain complexing agents (e.g. EDTA) increase colour intensity to aid detection and determination for example complexing [Fe(H2O)6]3+(aq) ions with colourless thiocyanate ions (SCN-) to produce the more deeply coloured [Fe(SCN)(H2O)5]2+(aq) complex ion which can be used to detect low concentrations of iron in substances like tea by comparing absorbance against a calibration curve of known concentrations. in a colorimeter interchangeable filters are used to illuminate the sample with its complementary colour where absorption is greatest hence sensitivity optimised it is also possible to determine the formula of a complex ion as the complexing agent is added to separate batches of the transition metal sample the intensity of the colour will increase until there is no more transition metal ions for it to combine with (the volume would be kept constant using water) this allows us to determine the number of complex ions that combine with a transition metal where both concentrations are known (ideally the same value) Summary Questions Exam Style Questions Page 222 Page 233 A2 Chemistry AQA (Nelson Thornes) Chemguide 1, 2 3 (if not already done) 220 - 222 Colour LUND 7 May 2017 35 A2 Unit 5 Energetics, Redox and Inorganic Chemistry Variable Oxidation States across the d block the effective nuclear charge increases hence relative stability of 2+ oxidation state cf 3+ increases as the e- are better held as the number of 4s/3d electrons increases from Ti to Mn so does the maximum oxidation state (Sc=3, Ti = 4, V = 5, Cr = 6, Mn = 7). thereafter the maximum declines as effective nuclear charge increases suggesting that the 4s and unpaired 3d electrons only are involved transition metals with charges > +3 cannot exist in aqueous solution where they exist as oxoanions instead e.g. MnO4-, Cr2O72-(aq), CrO42-(aq) with covalent bonding between the oxygen and the transition metal (can you suggest their shape?) this can be explained in two ways – a lot of energy would be required to form a 4+ ion, and if it existed it would have a large charge density (thus be highly polarising) so would react with water molecules and decompose them +2 state tends to be reducing, as exemplified by Fe2+ in the manganate(VII) titration some +2 ions are unstable in air due to aerial oxidation (where they are themselves reducing agents) this can be pH dependent and occurs more readily in alkaline conditions e.g. keeping [Fe(H2O)6]2+(aq) in acidic solution helps it resist aerial oxidation to [Fe(H2O)6]3+(aq) Redox Titrations higher oxidation states (typically +4 and higher) are good oxidising agents MnO4-(aq) and Cr2O72-(aq) are particularly good as oxidising agents in redox titrations you will need to balance redox equations – some revision may be necessary here the titration procedure is pretty much the same as with acid-base titrations acidic conditions are employed and you should be able to carry out a calculation to determine the minimum amount of sulphuric acid required sulphuric acid is preferred (can you explain why each of the following: hydrochloric, nitric and ethanoic might not be suitable?) Manganate(VII) shows a distinct colour change whilst dichromate(VI) require an indicator since both Cr2O72-(aq) and [Cr(H2O)6]3+ ions are coloured the indicator used is sodium N-phenylamine-4-sulphonate which turns from colourless to purple at the end point in case you wondered it works by changing colour at a particular electrode potential, in this case + 0.84V Summary Questions Exam Style Questions Page 228 Page 233 A2 Chemistry AQA (Nelson Thornes) Chemguide 2, 3 1(some extra reading will be needed) 223 - 226 Variable oxidation state, redox titration LUND 7 May 2017 36 A2 Unit 5 Energetics, Redox and Inorganic Chemistry Chromium The oxidation of cobalt is covered later on Reducing Chromium (VI) to Chromium(III) chromium in chromate(VI) can be reduced by reacting it with Zn in the presence of conc. HCl (this also releases H2 - a reducing atmosphere) the feasibility can be demonstrated using electrode potentials Cr2O72-(aq) + 14H+(aq) + 6e- 2Cr3+(aq) + 7H2O(l) orange green 3+ Cr (aq) + e Cr2+(aq) green blue Zn2+(aq) + 2e Zn(s) Eo +1.33V -0.41V -0.76V the +2 state is readily oxidised back to the +3 state by air – unless preserved in a reducing (e.g. H2) atmosphere Ox State +6 +3 +2 Chromium CrrO7 orange CrO42- (shape?) yellow 3+ [Cr(H2O)6] Green 2+ [Cr(H2O)6] Blue 2- Oxidising Chromium (III) to Chromium(VI) oxidation of transition metals tends to occur more readily in alkaline conditions Iron(II) sulphate for example is kept in acidic conditions to prevent aerial oxidation a plausible reason is that it is harder to remove electrons from the positively charged complex present in acidic solutions Cr3+ can be oxidised to chromate(VI) by H2O2 in strongly alkaline conditions initially further deprotonation in xs OH- produces deep green [Cr(OH)6]3-(aq) [Cr(H2O)6]3+(aq) + 6OH(aq)- [Cr(OH)6]3-(aq) + subsequent oxidation with H2O2 yields yellow chromate(VI) CrO42-(aq) tetraoxochromate(VI)) ions) LUND 7 May 2017 37 6H2O(l) ions (aka A2 Unit 5 Energetics, Redox and Inorganic Chemistry writing half equations for redox reactions under alkaline conditions is a little more tricky than under acidic conditions – but here is a useful ‘cheat’ do it exactly as if it was in acidic conditions then cancel out the hydrogen ions by adding hydroxide ions equally to both sides i.e. H2O2(aq) + 2H+(aq) + 2e- 2OH-(aq) 2H2O(l) cancelling 2H+, and subsequently H2O [Cr(OH)6]3-(aq) H2O2(aq) + 2e- CrO42-(aq) + 2H2O(l) + 2H+(aq) + 3e- CrO42-(aq) + 4H2O(l) cancelling 2H+, and subsequently H2O [Cr(OH)6]3-(aq) + 2OH-(aq) +3e- now balancing for electrons and combining: 2[Cr(OH)6]3-(aq) + 3H2O2(aq) 2CrO42-(aq) + 2OH-(aq) + 8H2O(l) upon acidification orange dichromate(VI) Cr2O72-(aq) is formed – this is an acid-base equilibrium NOT a redox – check the oxidation state of chromium (you should be able to write the equation) see if you can write half equations and then a full balanced equation for other oxidations carried out in alkaline conditions e.g.: [Co(NH3)6]2+(aq) to [Co(NH3)6]3+(aq) by aerial oxygen Co(OH)2(s) oxidised by H2O2 to Co(OH)3(s). Fe(OH)2(s) to Fe(OH)3(s) by aerial oxygen Summary Questions Page 228 A2 Chemistry AQA (Nelson Thornes) Chemguide 1, 3 204 – 7, 226 – 228, 238 Chromium LUND 7 May 2017 38 A2 Unit 5 Energetics, Redox and Inorganic Chemistry Catalysis by Transition Metals catalysts enable a different mechanism with a different activation energy (hence different rate) whilst being chemically unchanged at the end of a reaction NOTE CATALYSTS CHANGE THE VALUE EA NOT THE SHAPE OF THE CURVE. Heterogeneous Catalysis heterogeneous catalysis – the catalyst is in a different phase to the reactants typically transition metals or their compounds are used e.g. manufacture of ammonia Haber Process Fe catalytic converters Pt and Rh hardening fats (making margarine) Hydrogenation Ni (adsorption onto the surface of the solid nickel catalyst weakens π bonds) manufacture of nitric acid Ostwald Process Pt and Rh manufacture of sulphuric acid Contact Process V2O5 adsorption occurs onto active sites and consequently: weakens the bonds in the reactants hence lowers the activation energy improves the stereochemistry for collisions by orienting molecules favourably provides a localised relatively high concentration of reactants LUND 7 May 2017 39 A2 Unit 5 Energetics, Redox and Inorganic Chemistry adsorption must be strong enough to hold the reactant for long enough to promote a reaction but must not be too strong (e.g. as with tungsten) otherwise regeneration of active sites is too slow as the product undergoes desorption from the surface in general the strength of adsorption decreases from left to right in the transition metals and the desorption occurs more readily in the same direction hence Fe (Haber process) Co and Ni (Hydrogenation) are commonly used since they offer a compromise Catalytic Converters platinum and rhodium are coated onto a honeycomb ceramic material (minimises costs whilst providing a large surface area = increased rate) since adsorption only occurs at the surface (expensive metal underneath would be wasted) the reactant gases form weak bonds with the surface of the catalyst (adsorption) this weakens their bonds thus lowering the activation energy (additionally the catalyst also helps promote more favourable molecular orientation) this is followed by desorption in which the products depart the catalyst selected provides bonding strong enough to hold the reactant gases on the surface whilst not preventing the products from leaving thus blocking an active site Write equations for these CO and NO react to form CO2 and N2 NO also reacts with uncombusted hydrocarbons to produce CO2, H2O and N2 poisoning can occur if impurities contaminate the active sites e.g. sulphur dioxide and lead poisons catalytic converters hence unleaded low sulphur fuels must be used in addition the finely coated Pt/Rh can be lost from the surface this reduces efficiency and can result in an MOT failure and a large bill Haber process Pea sized Fe lumps are the catalyst - large surface area (enhanced by an aluminium oxide promoter) to increase rate without requiring an even higher temperature (energy cost plus unfavourable for yield) the iron catalyst does not effect the equilibrium position as both forward and backward reactions are favoured equally sulphur impurities (present in natural gas) can poison the iron so ‘scrubbing’ is carried out to remove the sulphur compounds (carbon monoxide can also be a problem) however it eventually has to be replaced LUND 7 May 2017 40 A2 Unit 5 Energetics, Redox and Inorganic Chemistry Methanol Production methanol is used as a chemical feedstock and as an additive to petrol it can be manufactured by the reversible reaction between carbon monoxide and hydrogen in the presence of a copper catalyst or alternatively Cr2O3 the reactants (‘synthesis gas’) are manufactured from the reaction of methane or propane with steam Contact process V2O5 used rather than faster Pt as lowers costs and less prone to poisoning specific use of the variable oxidation states of transition metals is made +5 V2O5(s) +4 + SO2(g) + +4 V2O4(s) 1 O2(g) 2 + V2O4(s) + SO3(g) +5 V2O5(s) Reactants Product Homogeneous Catalysis homogeneous catalysis – the catalyst is in the same phase to the reactants in this case the reaction proceeds via an intermediate species and will typically have a two step reaction profile with two activation energies both less than that for the uncatalysed reaction same phase as reactants (e.g. all in solution): acid catalysed esterification enzymes in biological systems chlorine free radicals (formed by the action of UV light on CFC’s) and ozone (O3) depletion Peroxodisulphate and iodide ions redox reaction between peroxodisulphate (S2O82-) and iodide ions is slow as both are negatively charged catalysed by iron(II) (or iron(III) – either will do) – note oppositely charged ions now react +2 Fe2+(aq) +3 + S2O82-(aq) + 2I-(aq) 2SO42-(aq) + +3 2Fe3+(aq) +2 I2(aq) Reactants Fe3+(aq) Products this could be followed experimentally using a colorimeter LUND 7 May 2017 41 + Fe2+(aq) A2 Unit 5 Energetics, Redox and Inorganic Chemistry Autocatalysis some reactions can speed up rather than slow down relative to the initial rate there can be several reasons for this: an oxide layer on a surface is being removed before the acid gets at the metal the reaction might be exothermic the product is itself a catalyst for the reaction – this is autocatalysis Conc. Slow reduction in conc. initially Faster reduction in conc. as autocatalysis begins Reaction slows down as reagents run out time in all cases the reaction will eventually start to slow down as reactants are used up e.g. manganate(VII) initially reacts slowly with ethanedioate ions (from oxalic acid) 2MnO4-(aq) + 5C2O42-(aq) 2Mn2+(aq) + 10CO2(g) + 8H2O(l) the Mn2+ ions produced autocatalyse the reaction, hence it actually speeds up once started they change to Mn3+ initially but are changed back in the next step: MnO4-(aq) + 16H+(aq) + 4Mn2+(aq) + 8H+(aq) 5Mn3+(aq) + 4H2O(l) 2Mn3+(aq) + C2O42-(aq) 2Mn2+(aq) + 2CO2(g) this could be followed experimentally using a colorimeter Summary Questions Exam Style Questions Exam Style Questions Page 232 Page 233 Page 247 A2 Chemistry AQA (Nelson Thornes) Chemguide 1-4 1, 2, 4 4, 8 229 - 230 Heterogeneous catalysis LUND 7 May 2017 42 A2 Unit 5 Energetics, Redox and Inorganic Chemistry Applications of Transition Metal Complexes EDTA this is an ethylenediamminetetraacetate ion or EDTA for short EDTA complexes are very stable – in effect a protective cage is formed around the transition metal ion thus isolating it from a biological system the replacement of unidentate with multidentate ligands is favoured by entropy since the total number of particles increases (see the section on entropy) Uses of EDTA 1. 2. 3. 4. antidote to Hg/Pb poisoning (traps metal ions) Ca2+ trap in blood transfusions – prevents clotting removal of Ca2+ from hard water (e.g. in shampoo) titimetric determination of metal ion concentration Haemoglobin haem – forms four co-ordinate bonds (tetradentate) with Fe2+ (a porphyrin structure) N in globin – a protein forms a fifth to form haemoglobin O2 or H2O form the sixth bond in oxyhaemoglobin or deoxyhaemoglobin as oxygen is a poor ligand it is easily released in cells lack of iron in the blood can cause anaemia as insufficient oxygen is transported resulting in tiredness and fatigue (or is that homework) taking iron tablets which contain soluble iron(II) sulphate counteracts this CO (which is a better ligand than oxygen) bonds with haemoglobin more strongly to form the relatively stable carboxyhaemoglobin thus reducing the bloods capacity to transport oxygen cyanide ions act in a similar way similar structures are found in a range of biologically important substances such as vitamin B12 (cobalt), and chlorophyll (magnesium) LUND 7 May 2017 43 A2 Unit 5 Energetics, Redox and Inorganic Chemistry Cis-platin cis-platin - [Pt(NH3)2Cl2] - is used in treating certain cancers (note the DNA complexing explanation on page 219 is wrong as it’s the chlorines that are displaced) its full name is cis-diamminedichloroplatinum(II) in case you wondered it forms DNA cross links via the platinum which damage the cancer cells http://www.youtube.com/watch?v=Wq_up2uQRDo&feature=related however, it does have side effects as it also effects normal cells e.g. renal toxicity, bone marrow suppression (loss of white blood cells increases the risk of other infections), fatigue and hearing loss and can also induce nausea and vomiting. testing renal function, blood and hearing is recommended before each cycle of therapy. so a cautious approach to dosage is necessary http://www.cancerhelp.org.uk/about-cancer/treatment/cancer-drugs/cisplatin geometrical isomerism possible the other form being trans-platin (which has no effect on cancer for stereochemical reasons Tollens’ Reagent diamminesilver(I) ion [Ag(NH3)2]+(aq) formed in ammonical silver nitrate (Tollens’ reagent) used in silver mirror test for aldehydes and distinguish them from ketones [Ag(NH3)2]+(aq) is reduced to Ag – the silver mirror - and NH3 is displaced see if you can write a balanced redox equation under alkaline conditions complexing prevents the precipitation of Ag2O in alkaline conditions which would otherwise mask the test [Ag(NH3)2]+(aq) is also formed when testing for silver chloride and silver bromide with the addition of ammonia following the silver nitrate test the ligand displacement allows the precipitate to be re-solvated A2 Chemistry AQA (Nelson Thornes) Chemguide 69 – 70, 217 – 219, 241 Cis-platin, haemoglobin, edta, Tollens’ LUND 7 May 2017 44 A2 Unit 5 Energetics, Redox and Inorganic Chemistry Metal-aqua Ions in aqueous solution tri-positive and di-positive TM ions form hexaaqua- complexes ligands (electron pair donors) are Lewis bases and transition metal ions (electron pair acceptors) are Lewis acids the presence of the ligand creates the familiar colour of transition metal solutions this can also be locked into the crystalline structure – again imparting colour (.xH2O) e.g. white anhydrous copper(II) sulphate dissolves (very) exothermically in water to form blue hexaaquacopper(II) ions [Cu(H2O)6]2+(aq) [Co(H2O)6]2+(aq) [Fe(H2O)6]2+(aq) [Cr(H2O)6]3+(aq) [Fe(H2O)6]3+(aq) [Al(H2O)6]3+(aq) Blue Pink Green Green Yellow Colourless Actually they are lilac but the presence of a small amount of orange [Fe(H2O)5OH]2+(aq) makes it appear yellow (see later) Hydrolysis of Metal-aqua Ions water ligands have increased Oδ-H δ+ bond polarity which promotes the abstraction of a hydrogen ion by another water molecule compared to that which takes place in the autoionisation of water (see Kw) Note the charge ! [Fe(H2O)6]3+(aq) Pale Lilac ACID + [Fe(H2O)5OH]2+(aq) + H3O+(aq) Orange oxonium ion BASE ACID H2O(l) BASE this is hydrolysis (reaction with water) and makes the solution pH around 2 for a 1M solution it would be wrong to assume that most populous species is the complex ion on the RHS as the equilibria still lies strongly to the LHS, however it does result in an increased hydrogen ion concentration further deprotonation very limited as water is a relatively weak base and the charge on the complex ion is less positive and so the Oδ-H δ+ bond polarity is less pronounced. the solution appears yellow as the orange colour is more intense than the pale lilac relative acidity of M3+ cf M2+ reflects the relative polarising power of the central transition metal on the polarity of the O-H bond of the ligand the chemistry of Al3+(aq) is similar to tri-positive transition metal ions Summary Questions How science works Page 238 Page 243 Page 235 A2 Chemistry AQA (Nelson Thornes) Chemguide 1, 2 1 Theories of acids 234 - 7 Acidity of hexaaqua LUND 7 May 2017 45 A2 Unit 5 Energetics, Redox and Inorganic Chemistry Reactions of Transition Metal-aqua ions with OH-(aq) upon the addition of sodium hydroxide a metal hydroxide precipitation occurs Cr(OH)3(s) Fe(OH)3(s) Al(OH)3(s) green orange white Cu(OH)2(s) Co(OH)2(s) Fe(OH)2(s) pale blue blue green goes brown on standing turns orange on standing (some texts will show the water ligands as well) deprotonation reactions occur with the addition of basic hydroxide ions to a greater extent than in aqueous solution and this eventually presents a neutral complex ion and thus precipitation (hydroxide ions are a better base than water) alternatively this reaction can be depicted as a reaction between a hydroxide ion and the oxonium ion (based on the equation on page 45) with a consequential shift in equilibria – either is acceptable as the outcome is essentially the same, but direct abstraction by OH-(aq) is easier to produce equations for. [Cr(H2O)6]3+(aq) + OH-(aq) [Cr(H2O)5OH]2+(aq) + H2O(l) [Cr(H2O)5OH]2+(aq) + OH-(aq) [Cr(H2O)4(OH)2]+(aq) + H2O(l) [Cr(H2O)4(OH)2]+(aq) + OH-(aq) [Cr(H2O)3(OH)3](s) + H2O(l) here there is no repulsion since there is no charge and so the complexes can hydrogen bond together producing a gelatinous precipitate. all the hydroxide precipitates are solvated by the addition of acid which reverses the equilibria (equations must be known – acid + base salt + water). precipitates insoluble in XS sodium hydroxide solution, but soluble in acid, are basic Fe(OH)3(s) Cu(OH)2(s) Fe(OH)2(s) Co(OH)2(s) XS NaOH(aq) XS sodium hydroxide can cause the precipitate to re-dissolve for amphoteric hydroxides due to further deprotonation for: Cr(OH)3(s) Al(OH)3(s) [Cr(H2O)3(OH)3](s) + OH-(aq) [Cr(H2O)2(OH)4]-(aq) + H2O(l) the charged particle created can now be solvated and if the sodium hydroxide solution is concentrated enough then the hexahydroxo- complex can eventually be formed. [Cr(H2O)2(OH)4]-(aq) + OH-(aq) [Cr(H2O)(OH)5]2-(aq) + H2O(l) [Cr(H2O)(OH)5]2-(aq) + OH-(aq) [Cr(OH)6]3-(aq) LUND 7 May 2017 46 + H2O(l) Any one of these ions would be credited in the exam, but this one is easiest to remember. A2 Unit 5 Energetics, Redox and Inorganic Chemistry Reactions of Transition Metal-aqua Ions in Solution with CO32-(aq) carbonate ions react with oxonium ions to yield carbon dioxide gas CO32-(aq) + 2H3O+(aq) CO2(g) + 3H2O(l) the relatively high acidity of tri-positive transition metal results in their metal carbonates being unstable [Fe(H2O)6]3+(aq) + 2H3O+(aq) CO32-(aq) + [Fe(H2O)5OH]2+(aq) + H2O(l) CO2(g) + H3O+(aq) 3H2O(l) thus carbonate ions react with tri-positive transition metal ions to produce carbon dioxide gas HENCE FIZZING in addition to a hydroxide precipitate the deprotonation equilibrium is shifted to the RHS as the carbonate ion removes the oxonium ion until the neutral triaquatrihydroxo- complex (the precipitate) is obtained the overall equation should be known but can be derived on the basis of a shift in equilibrium as the carbonate ion reacts with oxonium ion (it is slightly harder to work it out starting with the carbonate ion abstracting a hydrogen ion directly, but is also acceptable) 2[M(H2O)6]3+(aq) + Cr(OH)3(s) green 3CO32-(aq) 2[M(H2O)3(OH)3](s) + 3CO2(g) + 3H2O(l) Fe(OH)3(s) orange Al(OH)3(s) as previously it is soluble in acid since hydroxides are bases it is not soluble in xs sodium carbonate solution as the concentration of hydroxide ions is relatively low so no further deprotonation occurs. di-positive transition metal carbonates are stable as the oxonium ion concentration is relatively low hence metal carbonate precipitates are produced and no CO2(g) is evolved CoCO3(s) mauve CuCO3(s) A2 Chemistry AQA (Nelson Thornes) Chemguide blue-green 237 - 8 aqua ions hydroxide, aqua ions carbonate LUND 7 May 2017 47 FeCO3(s) green A2 Unit 5 Energetics, Redox and Inorganic Chemistry Reactions of Transition Metal-aqua Ions in Solution with NH3(aq) ammonia undergoes hydrolysis with water although it is only a weak alkali as the equilibria is biased to the LHS the hydroxide ion concentration is thus much lower than with fully ionised NaOH(aq) NH3(aq) BASE + H2O(l) ACID NH4+(aq) ACID + OH-(aq) BASE as with sodium hydroxide a metal hydroxide precipitation can be assumed to occur by direct deprotonation by the hydroxide ion. alternatively this reaction can be depicted as a reaction between a hydroxide ion and the oxonium ion (based on the equation on page 45) with a consequential shift in equilibria or abstraction of a proton by an ammonia molecule – either is acceptable as the outcome is essentially the same, but direct abstraction by OH-(aq) is easier to produce equations for and offers commonality with sodium hydroxide – so less to remember. Cr(OH)3(s) Fe(OH)3(s) Al(OH)3(s) green orange white Cu(OH)2(s) Co(OH)2(s) Fe(OH)2(s) pale blue blue green goes brown on standing turns orange on standing (some texts will show the water ligands as well) XS ammonia causes to [Cu(H2O)4(OH)2](s) to re-dissolve [Cu(H2O)4(OH)2](s) + 4NH3(aq) pale blue [Cu(NH3)4(H2O)2]2+(aq) deep blue Note that two water molecules remain + 2H2O(l) + 2OH-(aq) unlike adding xs NaOH further deprotonation is NOT an explanation of why solvation occurs since the concentration of hydroxide ions in the weakly alkaline ammonia solution is inadequate to further deprotonate the uncharged species with the far less polar Oδ-H δ+ bond polarity however, ammonia is a better ligand than hydroxide ions and water as its lone pair are less well held than for the relatively more electronegative oxygen hence ligand displacement can occur once the concentration of ammonia is high enough to bias the equilibria adequately the geometry is still octahedral as the ammonia ligand is roughly the same size as the water ligand – but is slightly distorted as the Cu-O bonds are weaker (water is a poorer ligand) therefore longer LUND 7 May 2017 48 A2 Unit 5 Energetics, Redox and Inorganic Chemistry similarly: [Co(H2O)4(OH)2](s) + 6NH3(aq) blue [Co(NH3)6]2+(aq) + 4H2O(l) + 2OH-(aq) pale yellow darkens on standing due to aerial oxidation to brown [Co(NH3)6]3+(aq) [Cr(H2O)3(OH)3](s) green [Cr(NH3)6]3+(aq) purple + 6NH3(aq) + 3H2O(l) + 3OH-(aq) aluminium, iron(II) and (III) DO NOT undergo these ligand displacement reactions hence their hydroxide precipitates are insoluble in xs ammonia Summary Questions Exam Style Questions Page 243 Page 244-5 A2 Chemistry AQA (Nelson Thornes) Chemguide 3, 4 2, 4, 5 239 – 240, 242 - 243 aqua ions ammonia Ligand Displacement Reactions (summary) replacement is stepwise and is reversible – this also means that it may be incomplete e.g. in the example below whilst [Cu(Cl)4]2-(aq) is yellow what will be seen is a green solution, implying that complete substitution has not occurred some ligands form more stable complexes than others which will effect the equilibrium position for a given set of conditions there is a relationship between effectiveness as a ligand, base and nucleophile as all three are essentially related to the availability of a lone pair e.g. cyanide ions are better than water/O2 – see haemoglobin section substitution by bidentate and multidentate ligands are also favoured by an increase in entropy as there will be a consequential increase in the number of independent particles moles of EDTA used = moles of T.M. ion and is useful for the analysis of the concentration of a transition metal (by titration or colorimetry) LUND 7 May 2017 49 A2 Unit 5 Energetics, Redox and Inorganic Chemistry concentration and temperature will effect the position of equilibrium and hence the colour e.g. [Co(H2O)6]2+(aq) pink + 4Cl-(aq) [Co(Cl)4]2-(aq) + blue 6H2O(l) [Cu(H2O)6]2+(aq) blue + 4Cl-(aq) [Cu(Cl)4]2-(aq) + yellow 6H2O(l) adding concentrated HCl(aq) will shift the equilibrium to the RHS the forward reaction is endothermic and hence a temperature change will effect the colour the above equilibrium is the basis of cobalt chloride paper used as a chemical test for water (e.g. in the combustion of a hydrocarbon demonstration) the co-ordination number is changed as water is replaced by the far larger chloride ion – usually associated with a significant colour change when water ligands are replaced by the similar sized ligands, e.g. ammonia, the octahedral geometry is retained the stability of the +II oxidation state can be altered by the ligand present e.g. yellow [Co(NH3)6]2+(aq) is readily oxidised to brown [Co(NH3)6]3+(aq) by oxygen from the air whereas [Co(H2O)6]2+ (aq) is not. similarly alkaline conditions/hydroxyl ligands can also make oxidation easier e.g. Co(OH)2(s) is readily oxidised by H2O2 to brown Co(OH)3(s). Fe(OH)2(s) is readily oxidised by oxygen from air to Fe(OH)3(s) in case you wondered this is because the electrode potentials in your text book relate to the hexaaqua complex and changing the ligand will modify its value a similar change in relative electrode potentials occurs when alkaline rather than acidic conditions are employed – see the oxidation of chromium Summary Questions Exam Style Questions Page 241 Page 244 A2 Chemistry AQA (Nelson Thornes) Chemguide 1-4 1, 3 239 – 241, 242 – 243, 228 Ligand displacement, cobalt, iron hydroxide LUND 7 May 2017 50 A2 Unit 5 Energetics, Redox and Inorganic Chemistry THIS IS AVAILABLE IN COLOUR – See ‘Mr Lund’s Classes’ at READ CHEMISTRY Example Solution NaOH(aq) XS NaOH(aq) NH3(aq) XS NH3(aq) Na2CO3(aq) Other test Result Fe3+ Fe2(SO4)3 FeCl3 Yellow Orange-brown gelatinous ppt. Insoluble Orange-brown gelatinous ppt. Insoluble Orange-brown gelatinous ppt. KMnO4 (acidified in dilute H2SO4) No reaction occurs. Fe2+ FeSO4 Green Green ppt., then, slowly turning orange due to aerial [O] Insoluble Insoluble Dark green ppt. KMnO4 (acidified in dilute H2SO4) Purple MnO4decolourise dFe2+ Fe3+ Cu2+ CuSO4 Pale blue Pale blue ppt. Insoluble Green ppt., then, slowly turning orange due to aerial [O] Pale blue ppt. Soluble (deep blue solution) Blue-green ppt. Co2+ CoCl2 Pink Blue ppt. then, slowly turning brown Insoluble Blue ppt. Mauve ppt. Al3+ Al2(SO4)3 Colourless White ppt. Soluble (colourless solution) White gelatinous ppt. Soluble (yellow solution which darkens on standing due to aerial [O]) Insoluble Cr3+ CrCl3 Green Grey-green ppt. Soluble (green solution) Grey-green to grey-blue gelatinous ppt. Soluble (slightly) -in excess a violet or pink solution formed. Green ppt. ION White ppt. Please note that the above colours are illustrative (it should be obvious that variables beyond control are concentration, colour on computer screen, colour of printer !!!) LUND 7 May 2017 51 A2 Unit 5 Energetics, Redox and Inorganic Chemistry THIS IS AVAILABLE IN COLOUR – See ‘Mr Lund’s Classes’ at READ CHEMISTRY Colour Cr Mn Fe Co Cu (all hexaaqua unless otherwise) Mn2+ (almost) Colourless Lilac/Purple Pink Blue Green Yellow Orange/Brown [Cr(NH3)6]2+ MnO4- CoCO3 [CoCl4]2- Cr2+ Co(OH)2 Cu2+ Cu(OH)2 [Cu(NH3)4]2+ CuCO3 [Co(NH3)6]2+ [CuCl4]2- Cr3+ Cr(OH)3 [Cr(OH)6]3CrO42_ Fe2+ Fe(OH)2 FeCO3 Fe3+ Cr2O72_ Fe(OH)3 Co(OH)3 [Co(NH3)6]3+ [Fe(SCN)]2+ Co2+ Red LUND 7 May 2017 52 Cu2O A2 Unit 5 Energetics, Redox and Inorganic Chemistry LUND 7 May 2017 53 A2 Unit 5 Energetics, Redox and Inorganic Chemistry A2 Module 5 Thermodynamics and Further Inorganic Chemistry Thermodynamics Enthalpy change (ΔH) be able to define and apply the terms enthalpy of formation, ionisation enthalpy, enthalpy of atomisation of an element and of a compound, bond dissociation enthalpy, electron affinity, lattice enthalpy (defined as either lattice dissociation or lattice formation), enthalpy of hydration and enthalpy of solution. be able to construct a Born.Haber cycle to calculate lattice enthalpies from experimental data be able to calculate enthalpies of solution for ionic compounds from lattice enthalpies and enthalpies of hydration be able to use mean bond enthalpies to calculate an approximate value of ΔH for other reactions be able to explain why values from mean bond enthalpy calculations differ from those determined from enthalpy cycles Free-energy change (ΔG) and entropy change (ΔS) understand that ΔH, whilst important, is not sufficient to explain spontaneous change (e.g. spontaneous endothermic reactions) understand that the concept of increasing disorder (entropy change ΔS) accounts for the above deficiency, illustrated by physical change (e.g. melting, evaporation) and chemical change (e.g. dissolution, evolution of CO2 from hydrogencarbonates with acid). be able to calculate entropy changes from absolute entropy values understand that the balance between entropy and enthalpy determines the feasibility of a reaction; know that this is given by the relationship ΔG = ΔH . TΔS be able to use this equation to determine how ΔG varies with temperature be able to use this relationship to determine the temperature at which a reaction is feasible Periodicity Study of the reactions of Period 3 elements Na . Ar to illustrate periodic trends be able to describe trends in the reactions of the elements with water, limited to Na and Mg. be able to describe the trends in the reactions of the elements Na, Mg, Al, Si, P and S with oxygen, limited to the formation of Na2O, MgO, Al2O3, SiO2, P4O10 and SO2. A survey of the acid-base properties of the oxides of Period 3 elements be able to understand the link between the physical properties of the highest oxides of the elements Na - S in terms of their structure and bonding. be able to describe the reactions of the oxides of the elements Na - S with water, limited to Na2O, MgO, Al2O3, SiO2, P4O10, SO2.and SO3. know the change in pH of the resulting solutions across the Period. be able to explain the trends in these properties in terms of the type of bonding present. be able to write equations for the reactions which occur between these oxides and given simple acids and bases. Redox Equilibria Redox equations be able to apply the electron transfer model of redox, including oxidation states and half equations to d-block elements Electrode potentials know the IUPAC convention for writing half-equations for electrode reactions. know and be able to use the conventional representation of cells. understand how cells are used to measure electrode potentials by reference to the standard hydrogen electrode know the importance of the conditions when measuring the electrode potential, Eo (Nernst equation not required). know that standard electrode potential, Eo , refers to conditions of 298 K, 100 kPa and 1.00 mol dm−3 solution of ions. Electrochemical series know that standard electrode potentials can be listed as an electrochemical series. be able to use Eo values to predict the direction of simple redox reactions and to calculate the e.m.f of a cell. LUND 7 May 2017 54 A2 Unit 5 Energetics, Redox and Inorganic Chemistry Electrochemical cells appreciate that electrochemical cells can be used as a commercial source of electrical energy appreciate that cells can be non-rechargeable (irreversible), rechargeable and fuel cells be able to use given electrode data to deduce the reactions occurring in non-rechargeable and rechargeable cells and to deduce the e.m.f. of a cell understand the electrode reactions of a hydrogen-oxygen fuel cell and appreciate that a fuel cell does not need to be electrically recharged appreciate the benefits and risks to society associated with the use of these cells Transition Metals General properties of transition metals know that transition metal characteristics of elements Ti - Cu arise from an incomplete d sub-level in atoms or ions. know that these characteristics include complex formation, formation of coloured ions, variable oxidation state and catalytic activity. Complex formation be able to define the term ligand be able to define the term ligand know that co-ordinate bonding is involved in complex formation. understand that a complex is a central metal ion surrounded by ligands. know the meaning of co-ordination number. understand that ligands can be unidentate (e.g. H 2O, NH3 and Cl−) or bidentate (e.g. NH2CH2CH2NH2 and C2O42-) or multidentate (e.g. EDTA4- ). know that haem is an iron(II) complex with a multidentate ligand. Shapes of complex ions know that transition metal ions commonly form octahedral complexes with small ligands (e.g. H 2O and NH3). know that transition metal ions commonly form tetrahedral complexes with larger ligands (e.g. Cl− ). Know that square planar complexes are also formed e.g. cisplatin know that Ag+ commonly forms linear complexes, (e.g. [Ag(NH3)2]+, [Ag(S2O3)2]3− and [Ag(CN)2]−). Formation of coloured ions know that transition metal ions can be identified by their colour, limited to the complexes in this module. know that colour changes arise from changes in oxidation state, co-ordination number and ligand. know that colour arises from electronic transitions from the ground state to excited states: ΔE = hv. appreciate that this absorption of visible light is used in spectrometry to determine the concentration of coloured ions Variable oxidation states know that transition elements show variable oxidation states. know that Cr3+ and Cr2+ are formed by reduction of Cr2O72- by zinc in acid solution. know the redox titrations of Fe2+ with MnO4- and Cr2O72− in acid solution. be able to perform calculations for these titrations and for others when the reductant and its oxidation product are given. know the oxidation of Co2+ by air in ammoniacal solution. know the oxidations in alkaline solution of Co2+ and Cr3+ by H2O2. Catalysis know that transition metals and their compounds can act as heterogeneous and homogeneous catalysts. Heterogeneous know that a heterogeneous catalyst is in a different phase from the reactants and that the reaction occurs at the surface. understand the use of a support medium to maximize the surface area and minimize the costs and that the reaction occurs at the surface understand how V2O5 actsas a catalyst in the Contact Process. know that a Cr2O3 catalyst is used in the manufacture of methanol from carbon monoxide and hydrogen. Know that Fe is used as a catalyst in the Haber process know that catalysts can become poisoned by impurities and consequently have reduced efficiency; know that this has a cost implication (e.g. poisoning by sulphur in the Haber Process and by lead in catalytic converters in cars) Homogeneous know that when catalysts and reactants are in the same phase, the reaction proceeds through an intermediate species (e.g. the reaction between I− and S2O82- catalysed by Fe2+ and autocatalysis by Mn2+ in titrations of C2O42- with MnO4−) LUND 7 May 2017 55 A2 Unit 5 Energetics, Redox and Inorganic Chemistry Other applications of transition metal complexes understand the importance of variable oxidation states in catalysis; both heterogeneous and homogeneous catalysts understand that Fe(II) in haemoglobin enables oxygen to be transported in the blood, and why CO is toxic know that the Pt(II) complex cisplatin is used as an anticancer drug understand that [Ag(NH3)2] + is used in Tollens’ reagent to distinguish between aldehydes and ketones Reactions of Inorganic Compounds in Aqueous Solution Lewis acids and bases know the definitions of a Lewis acid and Lewis base; understand the importance of lone pair electrons in co-ordinate bond formation. Metal-aqua ions know that metal.aqua ions are formed in aqueous solution: [M(H2O)6]2+, limited to M = Fe, Co and Cu [M(H2O)6]3+, limited to M = Al, Cr and Fe know that these aqua ions can be present in the solid state (e.g. FeSO 4.7H2O and Co(NO3)2.6H2O). Acidity or hydrolysis reactions understand the equilibria [M(H2O)6]2+ + H2O [M(H2O)5(OH)]+ + H3O+ and [M(H2O)6]3+ + H2O [M(H2O)5(OH)]2+ + H3O+ to show generation of acidic solutions with M3+, and very weakly acidic solutions with M2+. understand that the acidity of [M(H2O)6]3+ is greater than that of [M(H2O)6]2+ in terms of the polarising power (charge/size ratio) of the metal ion be able to describe and explain the simple test-tube reactions of M2+ (aq) ions, limited to M = Fe, Co and Cu, and of M 3+ (aq) ions, limited to M = Al, Cr and Fe, with the bases OH¯, NH3 and CO32−. know that MCO3 is formed but that M2(CO3)3 is not formed. know that some metal hydroxides show amphoteric character by dissolving in both acids and bases (e.g. hydroxides of Al 3+ and Cr3+). know the equilibrium reaction 2CrO42- + 2H+ ⇋ Cr2O72− + H2O Substitution reactions understand that the ligands NH3 and H2O are similar in size and are uncharged, and that ligand exchange occurs without change of co-ordination number (e.g. Co2+ and Cr3+). know that substitution may be incomplete (e.g. the formation of [Cu(NH3)4(H2O)2]2+). understand that the Cl− ligand is larger than these uncharged ligands and that ligand exchange can involve a change of co-ordination number (e.g. Co2+ and Cu2+). know that substitution of unidentate ligand with a bidentate or a multidentate ligand leads to a more stable complex. understand this chelate effect in terms of a positive entropy change in these reactions LUND 7 May 2017 56 AS Chemistry Unit 2 57 AS Chemistry Unit 2 58