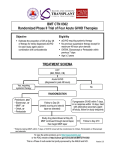
Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
PERSPECTIVE Disease Models & Mechanisms 4, 318-333 (2011) doi:10.1242/dmm.006668 Mouse models of graft-versus-host disease: advances and limitations Disease Models & Mechanisms DMM Mark A. Schroeder1 and John F. DiPersio1,* The limiting factor for successful hematopoietic stem cell transplantation (HSCT) is graft-versus-host disease (GvHD), a post-transplant disorder that results from immune-mediated attack of recipient tissue by donor T cells contained in the transplant. Mouse models of GvHD have provided important insights into the pathophysiology of this disease, which have helped to improve the success rate of HSCT in humans. The kinetics with which GvHD develops distinguishes acute from chronic GvHD, and it is clear from studies of mouse models of GvHD (and studies of human HSCT) that the pathophysiology of these two forms is also distinct. Mouse models also further the basic understanding of the immunological responses involved in GvHD pathology, such as antigen recognition and presentation, the involvement of the thymus and immune reconstitution after transplantation. In this Perspective, we provide an overview of currently available mouse models of acute and chronic GvHD, highlighting their benefits and limitations, and discuss research and clinical opportunities for the future. Introduction GvHD immunopathology in humans and mice A major complication and limitation to the broad application of hematopoietic stem cell transplantation (HSCT) is graft-versushost disease (GvHD), which results from immune-mediated attack of recipient tissue by donor T cells contained in the transplant. GvHD accounts for 15-30% of deaths that occur following allogeneic HSCT that is carried out to treat malignant diseases, and is a major cause of morbidity in up to 50% of transplant recipients (Ferrara et al., 2009). This high incidence is despite the use of aggressive immunosuppressive therapies after transplantation, as well as allele-level human leukocyte antigen (HLA) matching between donor and recipient. Despite recent advances to reduce the incidence of GvHD through altering prophylactic regimens and reducing the intensity of conditioning prior to transplantation, effective treatments for GvHD are lacking. Thus, accurate and reproducible experimental models of GvHD are essential for advancing our fundamental understanding of this disorder and for developing new treatments. In this Perspective, we provide an overview of available mouse models of acute and chronic GvHD, discuss their uses and limitations, and provide guidance on selecting an appropriate model. We also briefly discuss the graft-versus-leukemia (GVL) effect, although readers are referred to other recent reviews for more detail on this topic (Bleakley and Riddell, 2004; Ringden et al., 2009). We begin by describing the clinical symptoms and immunopathology of GvHD. In humans, GvHD manifests as acute GvHD (aGvHD), which occurs within 100 days of transplant, or as chronic GvHD (cGvHD), which typically develops 100 days after transplantation and can take 2-5 years to become clinically evident. This temporal distinction does not necessarily translate to mouse models of the disease, which can differ in time of onset and are mainly defined by their clinical phenotype: cGvHD develops within weeks after transplantation in most mouse models. In addition, in humans, aGvHD typically precedes cGvHD, although in some cases cGvHD can occur without the occurrence of clinically obvious aGvHD. One factor that confounds the translation of findings in mouse models to the human disease is that most patients are given immunosuppressive therapy to prevent aGvHD, and these medications might influence the development of cGvHD. Interestingly, interventions to prevent aGvHD in the clinic have, in general, not significantly decreased rates of cGvHD in humans (Lee, 2010). The sequence of events that lead to the development of GvHD has largely been defined using mouse models. Early work established that T-cell alloreactivity is the underlying cause of the disease (Korngold and Sprent, 1978; Sprent et al., 1986). The pathology of both acute and chronic mouse models of GvHD relies on T-cell alloreactivity, but each form has a different phenotype owing to differential involvement of cytotoxic (CD8+) or helper (CD4+) T-cell subsets. Donor CD8+ T cells are activated when their T-cell receptor (TCR) binds to recipient peptides presented in the context of recipient class I major histocompatibility complex (MHC) molecules (see Box 1 for a glossary of immunological terms). T-cell activation also requires the engagement of costimulatory receptors on activated donor T cells by their cognate ligands expressed on donor antigen-presenting cells (APCs). CD8+ T-cell cytotoxicity is mediated by perforin, granzymes and Fas ligand, and inflammatory cytokines augment this response. Donor CD4+ T cells are activated when their TCR binds to exogenous 1 Division of Oncology, Siteman Cancer Center, Washington University School of Medicine, St Louis, MO 63110, USA *Author for correspondence ([email protected]) © 2011. Published by The Company of Biologists Ltd This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial Share Alike License (http://creativecommons.org/licenses/bync-sa/3.0), which permits unrestricted non-commercial use, distribution and reproduction in any medium provided that the original work is properly cited and all further distributions of the work or adaptation are subject to the same Creative Commons License terms. 318 dmm.biologists.org PERSPECTIVE Mouse models of GvHD Disease Models & Mechanisms DMM Box 1. Glossary of immunological terms Antigen cross presentation: a process by which exogenous antigens derived from endogenous cells (such as damaged epithelium in the case of GvHD) are taken up by APCs and presented, typically in MHC class I molecules, to CD8+ T cells. In setting of GvHD, donor APCs cross-prime donor T cells by presenting recipient antigens. Antigen presentation: the process by which either self or non-self antigens are captured, processed and presented on the surface of APCs for cognate antigen recognition by lymphocytes. Antigen-presenting cells (APCs): a heterogenous population of cells (including B cells, monocytes/macrophages and dendritic cells) that present self and non-self antigens to T cells. Chimerism: the presence of two or more genetically distinct cell populations (from donor and recipient) in the same organism, as occurs following HSCT; can be measured by determining the percentage of donor cells present in the blood or bone marrow through genetic or immunocytometry testing. Cognate antigen recognition: recognition of antigen by T or B cells expressing receptors specific for that antigen. Cytokine storm: elevated levels of inflammatory mediators (such as TNF, IL-1 and IL-6) that potentiate further stimulation of immune effector cells such as T cells and macrophages in a positive-feedback loop; can occur in GvHD or in response to infections. Effector T cells: mature activated T cells that produce large amounts of cytokines (in the case of CD4+ T cells) or kill target cells via perforin and granzymes (in the case of CD8+ T cells). Epitope: a specific region of an antigen that is recognized by antibodies, or by B-cell or T-cell receptors. Epitope spreading: a normal or pathological (in the case of auto-antigens) immune response in which epitopes distinct from the inducing epitope become targets of the immune response. This typically occurs in the setting of chronic inflammatory responses. APCs process and present a variety of epitopes from the same larger antigenic peptide, resulting in an increased repertoire of T-cell responses against multiple epitopes from the original antigenic peptide. Major histocompatibility complex (MHC): a cluster of highly polymorphic genes that encode proteins involved in antigen presentation (also known as HLA antigens in humans and H2 in mice); important for enabling the immune system to differentiate between self and foreign tissues. MHC class I enables presentation of mainly endogenous antigens to CD8+ T cells, whereas MHC class II enables presentation of mainly exogenous antigens to CD4+ T cells. Minor histocompatibility antigens (miHAs): a class of polymorphic antigens thought to arise from polymorphisms in the genes encoding common cellular proteins; presented by MHC class I and are often responsible for organ rejection and GvHD in MHC-matched transplants. An example is the H-Y antigen. Regulatory T (TReg) cells: a subset of CD4+ T cells that is defined by expression of the transcription factor FoxP3 and that suppresses immune responses to a wide variety of antigens, including alloantigens and autologous antigens. T helper 1 (Th1) cell: a CD4+ T-cell effector subset characterized by the production of the cytokines IL-2, IFN and IL-12. Th1 cells can activate macrophages and B cells. T helper 2 (Th2) cell: a CD4+ T-cell effector subset characterized by the production of the cytokines IL-4, IL-5, IL-6 and IL-10. Th2 cells provide help to B cells for antibody production. T-cell alloreactivity: the T-cell response induced when a graft is performed with tissue of the same species but with a different genotype, owing to the recognition of foreign tissue (allograft) by the T cells. Th17 cells: a subset of CD4+ T effector cells that expresses proinflammatory molecules of the IL-17 family and mediates cytotoxic responses resulting in inflammation and tissue injury. Thymic negative selection (also known as central tolerance): the deletion of self-reactive T cells (i.e. T cells with autoimmune potential) during their development in the thymus. Disease Models & Mechanisms peptides presented in the context of class II MHC molecules on recipient APCs. CD4+ T-cell activation results in the development of a T-helper (Th)1 inflammatory response [marked by the production of interferon- (IFN), interleukin-12 (IL-12) and IL2] or a Th2 response (marked by the production of IL-4, IL-5, IL6 and IL-10). In mice, cGvHD mainly involves a Th2 response, whereas aGvHD is mainly Th1 biased. Cellular isolates used for HSCT in mice contain, among other cells, stem cells and T cells. Within 24 hours after transplantation, T cells traffic to secondary lymphoid organs (Panoskaltsis-Mortari et al., 2004; Beilhack et al., 2005). This initial migration of donor T cells and their interaction with recipient APCs is believed to play a crucial role in alloantigen priming and donor T-cell activation prior to egress to primary target organs (Kim et al., 2003; Beilhack et al., 2005). Activated donor T cells traffic and cause cytotoxicity in the gut, skin, liver, lung, thymus and lymph nodes. This unique tissue restriction is observed both in mouse models of GvHD and in human patients suffering from GvHD. Chemokines and adhesion molecules also play a crucial role in T-cell trafficking (Wysocki et al., 2005). aGvHD immunopathology aGvHD involves the direct cytotoxic effects of donor T cells on recipient tissues, the activation of APCs and an inflammatory cascade known as a ‘cytokine storm’, which involves the production of cytokines, including IL-1, IL-6, IL-12, IFN and tumor necrosis factor- (TNF). IFN has diverse roles, regulating immune suppression as well as cytotoxicity (for a review, see Lu and Waller, 2009). The impact of IFN on aGvHD is temporally related: IFN can have immunosuppressive effects if present immediately after transplant but inflammatory effects if it is added later (Brok et al., 1998). This inflammatory cascade contributes to a systemic syndrome of weight loss, diarrhea, skin changes and high mortality. Key events involved in the development of aGvHD in mice are illustrated in Fig. 1. First, priming of the immune response establishes a milieu for enhanced T-cell activation and expansion, and occurs as a direct result of ‘conditioning’. Conditioning suppresses the recipient’s immune system and is applied to allow engraftment of donor hematopoietic stem cells. It can be achieved with cytotoxic agents such as irradiation or chemotherapy, or with immunosuppressive agents such as anti-thymocyte globulin or drugs that target recipient T cells, such as fludarabine. The most common method of conditioning in mouse models is total body irradiation, which ablates the recipient’s bone marrow, allowing donor stem cell engraftment and preventing rejection of the graft by limiting the proliferation of recipient T cells in response to donor cells. Subsequent activation of donor T cells depends on cognate antigen recognition. T cells are also activated by non-cognate stimulation by cytokines such as IL-1 and TNF that are released during priming by many different cell types (Ferrara et al., 1993). Subsequent expansion of donor T cells and release of cytokines such as IFN leads to the activation of additional effector cell types, including neutrophils, monocytes and natural killer (NK) cells, which can result in further donor T-cell proliferation independent of their interactions with cognate antigen (Teshima et al., 2002). In addition, proinflammatory macrophages are activated, which can release IL-1, TNF and nitric oxide (Nestel et al., 1992; Cooke et al., 1998). 319 PERSPECTIVE Mouse models of GvHD A Immune priming Inflammatory cytokines Damage XRT Chemotherapy Damage to host tissues (skin, gut, liver) B TNFα IL-1 IL-6 T-cell activation and co-stimulation Lymph nodes and target organs Recipient APC Donor APC Endogenous antigen APC maturation and antigen presentation MH MHC II CD8+ T cell C cGvHD immunopathology Exogenous antigen MHC I CD4+ T cell Alloreactive T-cell expansion Disease Models & Mechanisms DMM Lymph nodes T-cell expansion D Trafficking Skin, gut, liver, lung CD4+ or CD8+ T cell Trafficking Blood ood and lymph lymp E Tissue injury and destruction Skin, gut, liver, lung Tissue damage Tissue-resident DC or macrophage TNFα IL-1 IFNγ TNFα IL-1 IFNγ Perforin/granzyme Fas/Fas-ligand Host cell Fig. 1. Steps to aGvHD in mice. The progression of events occurring in the development of aGvHD in the mouse is illustrated. The five crucial steps of immune priming (A), activation (B), T-cell expansion (C), T-cell trafficking (D) and host tissue injury (E) are outlined. DC, dendritic cell; XRT, radiation conditioning. See main text for full details. The final step leading to end organ damage in aGvHD is T-cellmediated tissue toxicity, which involves soluble mediators, including TNF, perforin, granzymes, Fas and Fas ligand (Baker et al., 1996; Braun et al., 1996; Graubert et al., 1997; Jiang et al., 2001; Maeda et al., 2005). In addition, T helper 17 (Th17) cells are a subset of T cells that have recently been shown to contribute to this final cytotoxic phase and can cause increased inflammation and cytotoxicity when supplemented in the graft in an experimental setting. A recent report demonstrated that Th17 cells were sufficient but not necessary to cause GvHD (Iclozan et al., 320 2009). Surprisingly, however, some studies have reported that their absence from the graft can also worsen GvHD (Yi et al., 2008; Carlson et al., 2009; Kappel et al., 2009). It is hoped that future studies will resolve this discrepancy to clarify the role of Th17 cells in GvHD pathology. A crucial counterpart to Th17 cells are regulatory T (TReg) cells, which have been shown to suppress a wide range of immune responses, including alloreactive and autoreactive T-cell responses (Edinger et al., 2003; Sakaguchi et al., 2006). Unlike aGvHD, cGvHD typically presents as an autoimmune-like syndrome. As well as effects mediated by T cells, cGvHD involves B-cell stimulation, autoantibody production and systemic fibrosis. Mouse models of cGvHD are associated with prolonged survival compared with mouse models of aGvHD. Additional clinical characteristics depend on the mouse model and can include splenomegaly and lymphadenopathy resulting from B-cell expansion, glomerulonephritis, biliary cirrhosis and scleroderma. The immunopathology underlying cGvHD is less well understood than that of aGvHD, in part because of the limitations of currently available mouse models. Unlike aGvHD, there is no unifying theory for the development of cGvHD, and the pathological mechanisms differ depending on the model system. Mouse models of cGvHD typically involve three main pathological mechanisms: autoantibody production, pro-fibrotic pathways and defective thymic function. Here, we describe the main events that underlie the phenotype, although models exploring the three main mechanisms will be discussed in detail later. A summary of our current knowledge of the presumed sequence of events leading to end organ damage in cGvHD is outlined in Fig. 2. First, unlike in aGvHD, immune priming in cGvHD models is not dependent on tissue damage from conditioning: compared with aGvHD models, the initial inflammatory milieu is reduced or absent in cGvHD models, which involve reduced levels of radiation or no conditioning at all. The outcome of transplantation is mixed T-cell chimerism in most models. Thymic damage can result in dysfunctional negative selection of autoreactive T cells, and there can be decreased TReg cell number or function. In addition, donor and recipient B cells recognize and process extracellular antigens for presentation in the context of MHC. All of these processes can contribute to the initiation of cGvHD. Second, unlike in aGvHD, T-cell activation is not dependent on recipient APCs, but is a mix of cognate antigen recognition and cross-priming of donor T cells. CD4+ cells are necessary and sufficient to cause cGvHD in mouse models, whereas cytotoxic CD8+ T cells are not required, in contrast to aGvHD (Rolink and Gleichmann, 1983; Rolink et al., 1983; Zhang et al., 2006). Recipient B cells are activated by minor antigens and Th2 cytokines, and mature to present processed antigens in the context of MHC to donor CD4+ T cells, which in turn co-stimulate B cells to produce autoantibodies and additional stimulatory cytokines (Morris et al., 1990a; Shimabukuro-Vornhagen et al., 2009). Third, T and B cells clonally expand in response to antigenic stimulation and costimulation, and form memory and effector cells. In addition, epitope spreading can occur, resulting in oligoclonal T-cell expansion. Fourth, clonally expanded T and B cells can traffic to target organ sites and mediate damage through cytokine-mediated dmm.biologists.org PERSPECTIVE Mouse models of GvHD A Immune priming Thymic damage Decreased number of TReg cells B-cell priming Antigen Antibody Thymus B Recipient and donor B cells Endogenous antigen Donor APC Exogenous antigen MHC I Mouse models of aGvHD B cell MHC MH II CD8+ T cell Disease Models & Mechanisms DMM B cell T-cell and an B-cell activation and co-stimulation Recipient APC C MHC II TReg cells Autoreactive T cell + Th2 cytokines (IL-4, IL-10) CD4+ T cell T-cell and B-cell expansion and autoantibody production Antigen-stimulated clonal T-cell expansion Epitope spreading CD4+ or CD8+ T cells B-cell clonal expansion Autoantibodies D Trafficking Skin, gut, liver, lung, salivary glands, oral mucosa Autoantibodies B cell d and lymph Blood Autoreactive T cell Autoactivated T cell Target organs E Tissue injury, inflammation and fibrosis Autoreactive T cell Cytokines (IL-13, IL-4, IL-10) Antigen recognition Cytokine receptor Fibroblasts + Direct cytotoxicity TGFβ Collagen Macrophage Host cell + B cell Autoantibodies PDGF receptor Fig. 2. Steps to cGvHD in mice. The progression of events occurring in the development of cGvHD in the mouse is illustrated. The five steps illustrate immune priming (A), activation of T and B cells (B), T-cell expansion and B-cell autoantibody production (C), trafficking to sites of tissue damage (D), and end organ damage through chronic inflammation and fibrosis (E). See main text for full details. Disease Models & Mechanisms fibrosis and chronic inflammation. Additionally, bone-marrowderived T cells inappropriately selected in a dysfunctional thymus, or through dysfunctional peripheral tolerance mechanisms, traffic to target organs, can be activated by self antigens and perpetuate the Th2 response. Finally, tissue injury results from activated T and B cells that release inflammatory and fibrosing cytokines, such as IL-13, IL-10 and IL-4, and activated macrophages producing transforming growth factor- (TGF) stimulate collagen production from fibroblasts, collectively leading to a sclerodermalike syndrome. Autoantibodies against double-stranded DNA, single-stranded DNA and chromatin can be deposited at the site of target organs, leading to immune-complex glomerulonephritis or causing activation of the platelet derived growth factor receptor, leading to fibrosis (Svegliati et al., 2007). Most mouse models of aGvHD (summarized in Table 1) involve the transplantation of T-cell-depleted bone marrow supplemented with varying numbers and phenotypic classes of donor lymphocytes (either splenocytes or lymph node T cells) into lethally irradiated recipients. The bone marrow provides donor stem cells that allow hematopoietic reconstitution after transplant; T-cell depletion is carried out to control the dose and type of immune cells that are delivered. The discovery of the MHC and minor histocompatibility antigens (miHAs) has been integral to advancing the field of HSCT. Differences in MHC antigens are largely responsible for immunemediated rejection of foreign tissues and cells; however, even in an MHC-matched setting, differences in miHAs can cause grafts to be slowly rejected. Unlike humans, inbred mouse strains are closely related with respect to MHC antigens (note that class I MHC is referred to as H2 in mice). Although two strains of mice might share the same H2 (i.e. they are MHC-I-matched), differences in miHAs can result in GvHD in the context of experimental HSCT. MHC-mismatched and miHA-mismatched models are discussed below. In addition, we describe more recently developed models involving the xenotransplantation of human cells into immunodeficient mice. The severity of aGvHD depends on several factors. First, the dose and type of T-cell subsets (i.e. CD4+, CD8+ or TReg cells) that are transferred with donor bone marrow can affect the severity of the disease (Sprent et al., 1988; Korngold, 1992; Edinger et al., 2003). Second, irradiation dose is proportional to the degree of tissue damage and the subsequent cytokine storm, and thus is directly proportional to aGvHD-related mortality in the mouse (Hill et al., 1997; Schwarte and Hoffmann, 2005; Schwarte et al., 2007). In addition, the dose of irradiation that is myeloablative varies by mouse strain: inbred strains are generally more susceptible to radiation than their F1 progeny, and different strains show susceptibility differences (e.g. BALB/c>C3H>C57Bl/6). Third, genetic disparities play a major role in disease. For example, even mice that are MHC matched can vary by miHAs that influence aGvHD severity. Fourth, variation in environmental pathogens between labs and in mice from different suppliers can affect GvHD (Nestel et al., 1992). Therefore, the same model of aGvHD can vary between investigators because of differences in radiation dose and delivery rate, differences in pathogens between animal facilities, and differences in T-cell dose and sources of T cells. 321 322 C57/Bl6 (H2b) B6C3F1 (H2k/b) B6D2F1 (H2b/d) B6AF1 (H2b/a) B10.BR (H2k) B6.C-H2bm1 (bm1) (H2b background with mutation at MHC I) B6.C-H2bm12 (bm12) (H2b background with mutation at MHC II) C3H/HeJ (H2k) C57/Bl6 (H2b) C57/Bl6 (H2b) C57/Bl6 (H2b) C57/Bl6 (H2b) C57/Bl6 (H2b) C57/Bl6 (H2b) DBA/2 (H2d) B10.D2 (H2d) Table 1 continued on next page. CBA or BALB.K (H2k) B10.BR (H2k) miHA mismatched BALB/c (H2d) Recipient strain C57/Bl6 (H2b) Donor strain MHC mismatched Table 1. Mouse models of aGvHD 800 cGy 750 cGy 950 cGy or sublethal XRT 950 cGy or sublethal XRT 750-900 cGy +/– Cytoxan 120 mg/kg No XRT No XRT or 1100-1300 cGy 600-1050 cGy 900 cGy 900 cGy Conditioning regimen miHA mismatches miHA mismatches MHC II mismatch MHC I mismatch Mismatched for MHC I, MHC II and miHAs Mismatched for MHC I, MHC II and miHAs Mismatched for MHC I, MHC II and miHAs Mismatched for MHCI, MHCII and miHAs Mismatched for MHCI, MHCII and miHAs Mismatched for MHCI, MHCII and miHAs Genetics Systemic disease; lethal 70 106 spleen cells and 1 107 TCD BM 4 106 TCD BM cells Systemic disease; lethal and 1 106 T cells, or by day 80 purified CD4+ or CD8+ T cells CD4+ (minor contribution by CD8+) Systemic disease if CD4+ T cells infused with death by 20-40 days 4 106 TCD BM and 15 106 CD4+ T cells 8 106 TCD BM cells Systemic disease; and 1 107 whole symptoms include tail splenocytes or scaling, ear erosions, 5 106 CD4+ or CD8+ diarrhea, hunching T cells Systemic disease if CD8+ T cells infused with death by 30-80 days Korngold and Sprent, 1987; Korngold, 1992 Korngold and Sprent, 1987; Korngold, 1992; Cooke et al., 1996; Kaplan et al., 2004; Fanning et al., 2009 Rolink et al., 1983; Sprent et al., 1986 Rolink et al., 1983; Sprent et al., 1986 Vallera et al., 1981; Blazar et al., 1991; PanoskaltsisMortari et al., 1998 2.5 105 to 2 107 TCD Systemic disease; lethal BM cells and 5depending on T-cell 25 106 spleen cells dose and source; or 0.5-1 106 lymph symptoms by 14-28 node cells days 4 106 TCD BM cells and 1.5–7.5 106 CD8+ T cells Ghayur et al., 1988; Nestel et al., 1992; Gendelman et al., 2004 Pickel and Hoffmann, 1977; Via et al., 1996a; Krenger et al., 2000a; Hildebrandt et al., 2004; Blaser et al., 2005; Niculescu et al., 2005 Kanamaru et al., 1984; Fowler et al., 1996 Blazar et al., 1991 van Leeuwen et al., 2002 Reference example(s) Systemic disease; lethal depending on T-cell dose Whole lymphoid cells (2-6 107) Systemic disease; lethal by 30 days if XRT used; symptoms in 30-50 days if no XRT Systemic disease by 1030 days; lethal 2 107 BM cells and 525 106 spleen cells or 0.1-1 106 lymph node cells Whole splenocytes: 110 107 cells if no XRT; 2-5 106 purified T cells with XRT Systemic disease by 1021 days; lethal Outcome Splenic T cells (0.52 106) and TCD donor BM cells Cell type and dose CD8+ CD4+ CD8+ CD4+ and CD8+ CD4+ and CD8+ CD4+ and CD8+ CD4+ and CD8+ CD4+ and CD8+ CD4+ and CD8+ Main T-cell type contributing to phenotype Disease Models & Mechanisms DMM PERSPECTIVE Mouse models of GvHD dmm.biologists.org Disease Models & Mechanisms BALB.b (H2b) BALB.b (H2 b) B10.D2 (H2d) B10 (H2b) C57/Bl6 (H2b) DBA/2 (H2d) NOD/SCID 2m-null Human peripheral blood T cells: naive or CD3/CD28 bead expanded Mismatched for MHC I, MHC II and miHAs Mismatched for MHC I, MHC II and miHAs Mistmatched for MHC I, MHC II and miHAs miHA mismatches miHA mismatches miHA mismatches miHA mismatches Genetics BM, bone marrow; cGy, centigray; RO, retro-orbital injection; TCD, T-cell depleted; XRT, radiation conditioning. 250-300 cGy 350 cGy BALB/cA-RAG2–/– IL2r Human PBMCs –/– No XRT or 200-250 cGy NOD/SCID IL2rγ-null (NSG) 820 cGy 850–1000 cGy 775 cGy Sub-lethal 600-750 cGy or Fractionated 1000 cGy Conditioning regimen Human PBMCs Xenotransplantation models BALB/c (H2d) Recipient strain B10.D2 (H2d) Donor strain miHA mismatched Table 1. Continued – CD4+ and CD8+ CD4+ CD8+ Berger et al., 1994; Zhang et al., 2005 Kaplan et al., 2004 Korngold and Sprent, 1987; Eyrich et al., 2005 Reference example(s) Systemic disease Systemic disease 1 107 human T cells RO injection Death from GvHD in 3050 days 5-30 106 1 107 or 5-10 106 Nervi et al., 2007 van Rijn et al., 2003 Ito et al., 2009; King et al., 2009 4 106 TCD BM cells Minimal systemic disease Korngold and Sprent, 1987 and 1 106 T cells, or with whole spleen cells but increased with purified CD4+ or + purified CD8+ T cells; CD8 T cells up to 50% mortality at day 25 Systemic disease 5 106 TCD BM cells and 2 106 T cells CD4+ Systemic disease; no cutaneous involvement 8 106 TCD BM cells and 107 unfractionated spleen cells or 1.4 106 CD4+ or 6 105 CD8+ T cells CD4+ (with some contribution from CD8+) Systemic disease Outcome 1 107 BM cells and 1 108 whole splenocytes Cell type and dose CD4 (minor contribution by CD8+) Main T-cell type contributing to phenotype Disease Models & Mechanisms DMM Mouse models of GvHD PERSPECTIVE 323 PERSPECTIVE Disease Models & Mechanisms DMM MHC-mismatched models The most commonly studied MHC-mismatched model of aGvHD is C57/Bl6 (H2b) r BALB/c (H2d) (transplantation of cellular isolates from C57/Bl6 (H2b) donors into BALB/c (H2d) recipients). MHC-mismatched models of aGvHD also contain a number of miHA mismatches, although the impact of miHA mismatches on the pathophysiology of the clinical phenotype observed is limited. All models incorporate myeloablative radiation conditioning either as a single dose or, when radiation doses of more than 1100 cGy are delivered, as a fractionated dose separated by 3-8 hours, which decreases gut toxicity. Most MHC-mismatched models are dependent on both CD8+ and CD4+ T cells. To dissect the mechanisms by which these T-cell subsets induce aGvHD, transgenic mice that have a mutant MHC I (which is involved in CD8+ T-cell activation) [B6.C-H2bm1 (bm1)] or MHC II (which is involved in CD4+ T-cell activation) [B6.C-H2bm12 (bm12)] have been used. The H2bm1 and H2bm12 transgenic mouse models have been important in dissecting the interaction of T cells with recipient and donor APCs (Sprent et al., 1990). CD8+ T cells induce aGvHD by engagement of the TCR and release of perforin, granzymes and Fas ligand (Via et al., 1996b; Graubert et al., 1997; Maeda et al., 2005). Only recipient APCs can stimulate CD8+ T cells in CD8+ T-cell-dependent models (Shlomchik et al., 1999). Conversely, either recipient or donor APCs can stimulate CD4+ T-cell responses (Anderson et al., 2005). TNF mediates the cytotoxic effects of alloreactive CD4+ T cells. The target of these cytotoxic T-cell responses is recipient epithelium, although the epithelium is not required to present antigen to T cells (Teshima et al., 2002). Another MHC-mismatched model of aGvHD is the ‘parent-toF1’ model. Parent-to-F1 transplants have been studied using either lethal irradiation regimens or no irradiation conditioning. The latter model allows the study of aGvHD without the effects of radiation, thus mimicking the situation in humans that undergo nonmyeloablative or ‘reduced intensity conditioning’ regimens. The cells transplanted into these models are either whole splenocytes (in the non-irradiated models) or purified T cells with T-celldepleted bone marrow (in the irradiated models). The development of aGvHD in these models depends on differences in class I and class II MHC antigens, and involves the effects of CD4+ and CD8+ T cells (Hakim et al., 1991). The characteristics of aGvHD differ in the irradiated and non-irradiated parent-to-F1 model: whereas the irradiated model exhibits weight loss and skin problems, the non-irradiated model has relatively limited skin pathology or weight loss, and aGvHD mainly manifests as immune deficiency and mixed chimerism, with lower mortality. Importantly, not all parental donor strains cause aGvHD. Some can cause cGvHD (e.g. DBA/2 r B6D2F1), possibly because of the natural bias of some strains to mount either a Th1- or Th2-cell response (De Wit et al., 1993; Nikolic et al., 2000; Tawara et al., 2008). miHA-mismatched models miHA-mismatched models of aGvHD represent human HSCT more closely than MHC-mismatched models, because MHCmismatched transplants in humans are not commonly performed. Similar to the human setting, miHA-mismatched models exhibit less morbidity than MHC-mismatched models, but still result in lethal aGvHD. An example of a miHA-mismatched transplant 324 Mouse models of GvHD model is B10.D2 (H2d) r BALB/c or DBA/2 (H2d). The aGvHD that occurs in this model is primarily dependent on CD4+ T cells and less on CD8+ T cells (Korngold and Sprent, 1987). However, transplants between congenic strains illustrate that differences in MHC (which means that alloantigens are presented differently in vivo) influence the type and specificity of the T-cell response underlying the GvHD phenotype (Kaplan et al., 2004). For example, in other miHA-mismatched transplant models, such as C3H.SW r Bl6 (H2b) and DBA/2 r B10.D2 (H2d), GvHD depends primarily on CD8+ T cells. Xenogeneic transplant models These models have been developed to generate a system whereby human T-cell-mediated aGvHD can be studied and manipulated in vivo. However, transplanting across species (xenotransplantation) requires significant immunosuppression to prevent graft rejection, because the immune response to xenografts is typically much more robust than to allografts. Initial attempts to xenotransplant human cells into mice were conducted using non-obese diabetic severe combined immunodeficiency (NOD/SCID) mice and resulted in a 1-20% rate of engraftment (Mosier et al., 1988; Hoffmann-Fezer et al., 1993; Christianson et al., 1997). Low engraftment was due to the fact that NOD/SCID mice lack functional T and B cells but retain NK cell function and can therefore respond to foreign antigens. Subsequently, better engraftment of xenotransplanted cells was obtained using RAG2-deficient and IL-2-receptor-deficient mice (BALB/cA-RAG2–/– IL2r–/–), which lack functional T, B and NK cells (van Rijn et al., 2003). Our group has used the NOD/SCID 2-microglobulin-null mouse (Christianson et al., 1997; Nervi et al., 2007) to image the trafficking of xenotransplanted human T cells in vivo. Using this system, we demonstrated that retro-orbital injection of human T cells into sublethally irradiated NOD/SCID 2m-null recipients resulted in consistent engraftment and expansion of human T cells, the trafficking of T cells to target organs including the lung, skin, liver and kidney, and clinical symptoms consistent with severe aGvHD. T cells could be detected 1 hour after retro-orbital injection and trafficked to secondary lymphoid organs, resulting in lethal GvHD; by contrast, intravenous injection of T cells caused them to traffic first to the lungs, and was associated with the development of less severe aGvHD and less morbidity and mortality (Nervi et al., 2007). The most permissive mouse model for human stem-cell and Tcell engraftment was derived by back-crossing mice carrying a homozygous deletion of the common -chain of the IL-2 receptor onto a NOD/SCID background [referred to as NOD/SCID IL2rnull (NSG) mice]. NSG mice lack T, B and NK cells, and also have reduced function of macrophages and dendritic cells. Transplantation of human peripheral blood mononuclear cells (PBMCs) causes an aGvHD-like syndrome that results in death by 20-50 days, and is manifested by overt human T-cell infiltration of mouse skin, liver, intestine, lungs and kidneys (Ito et al., 2009). Xenogeneic transplants of human PBMCs into immunodeficient mice require human APCs to process mouse antigens and present them in the presence of class II MHC. Because T-cell recognition of MHC molecules is restricted by species, human TCRs do not recognize mouse MHC, making this a primarily CD4+ T-celldependent model, at least in the case of NOD/SCID (Lucas et al., 1990). dmm.biologists.org PERSPECTIVE Mouse models of GvHD Disease Models & Mechanisms DMM Antigen-specific transgenic TCR models A major limitation of all MHC- and miHA-mismatched transplant models of aGvHD is that it is difficult to determine the specific alloantigen that the responding T cells recognize. In addition, the cytokine storm induced by conditioning can lead to antigenindependent bystander activation of T cells. One solution to this problem has been to use mouse strains that have T cells expressing TCRs of defined specificity (TCR-transgenic mice); these strains were generated initially to study thymic selection, but have more recently been useful for understanding T-cell alloresponses in GvHD. The specificity of the T cells in a TCR-transgenic mouse is generally restricted to a single peptide epitope that is either constitutively expressed by the transplanted allogeneic APCs or is administered as an exogenous antigen to stimulate a T-cell response. Examples of TCR-transgenic mice include H-Y, TEa, 2C, TS1, D011.10 and OT-II (Kisielow et al., 1988; Sha et al., 1988; Murphy et al., 1990; Grubin et al., 1997; Barnden et al., 1998; Wilkinson et al., 2009). These mice have been used to investigate several aspects of GvHD, including the role of antigen affinity (Yu et al., 2006), antigen-induced T-cell activation and proliferation, TReg cell function (Albert et al., 2005; Damdinsuren et al., 2010) and, more recently, cross presentation of antigens (Wang et al., 2009). resulting in the sclerodermatous changes (Hamilton, 1987; Korngold and Sprent, 1987). In addition, other cellular mediators have also been implicated: macrophages are responsible for TGF production, whereas other cell types such as eosinophils and mast cells have been implicated in the pathogenesis of fibrosis in target organs. Mediators secreted by eosinophils and mast cells can cause fibroblast proliferation and collagen production (Levi-Schaffer and Weg, 1997). Two examples of sclerodermatous models are B10.D2 r BALB/c and LP/J r Bl6, both of which require total body irradiation conditioning (Jaffee and Claman, 1983; DeClerck et al., 1986). The dose of radiation can also affect the severity of disease (Jaffee and Claman, 1983). The sclerodermatous cGvHD models provide important insights into the pathogenesis of fibrosis that occurs in cGvHD; however, there are a number of limitations of these models. First, the fibrosis in human cGvHD can be systemic or pleiotropic, unlike mouse models, which mainly involve the skin, suggesting other mechanisms might be at play in the development of systemic sclerosis. Second, the time course of development of skin changes is less rapid in humans than in mouse models. Finally, these mouse models fail to reproduce the full clinical spectrum seen in humans, which can involve target organs including the intestine, lungs, liver, salivary glands, eyes, oral mucosa, muscles, tendons and nerves. Mouse models of cGvHD Although the clinical phenotype and kinetics of cGvHD are distinct from aGvHD, with fibrosis and chronic inflammation predominating in a manner resembling autoimmune disease, most of the same organs are affected. There are five factors that can contribute to the pathology of cGvHD in humans and mouse models after HSCT: cytokine-mediated fibrosis, defective negative selection of T cells, deficiency of TReg cells, genetic polymorphisms and dysregulated B-cell responses. However, no mouse model developed thus far recapitulates all of these characteristics or exactly mimics what is seen in human cGvHD. Current mouse models of cGvHD (summarized in Table 2) can be broadly divided into sclerodermatous (pro-fibrotic) models, autoantibody-mediated (lupus-like) models and a more recently reported model in which thymic function is defective (Sakoda et al., 2007; Chu and Gress, 2008). The distinctions in the phenotypes of models of cGvHD and aGvHD result from differences in the factors underlying the pathogenesis of each model. Sclerodermatous models Sclerodermatous (pro-fibrotic) cGvHD models are characterized by fibrotic changes in the dermis, which can involve the lung, liver and salivary glands (Jaffee and Claman, 1983; Claman et al., 1985; Howell et al., 1989; McCormick et al., 1999; Levy et al., 2000). Transplantation results in full donor chimerism, and the resulting phenotype resembles scleroderma in humans, which occurs in up to 15% of patients with cGvHD (Skert et al., 2006). However, fibrosis in the mouse models begins within 30 days of transplantation, which is atypical of human cGvHD, which occurs months to years after transplantation. These models are MHC matched but differ in numerous miHAs. The pathology is dependent on CD4+ T cells that release Th2 cytokines, which can stimulate other cells to release fibrosing cytokines (such as IL-13 and TGF) (for a review, see Wynn, 2004), Disease Models & Mechanisms Autoantibody-mediated models Autoantibody-mediated (lupus-like) cGvHD models, which exhibit a pathology similar to that of lupus, are manifested by nephritis, biliary cirrhosis, salivary gland fibrosis, lymphadenopathy, splenomegaly, autoantibody production and, to a lesser extent, skin pathology. These models are MHC mismatched and classically involve parent-to-F1 transplants that result in mixed chimerism. The most commonly studied autoantibody model is the DBA/2 r B6D2F1 model. This model results in expansion of recipient B cells, leading to lymphadenopathy, splenomegally and autoantibody production. Both MHC I and II are mismatched in these models, but the pathology is dependent on donor CD4+ T cells (Rolink and Gleichmann, 1983; Rolink et al., 1983; Hakim et al., 1991) and is associated with the production of Th2 cytokines. Manipulating the cytokine balance by skewing the CD4+ T-cell response to a Th1 phenotype, by exposure to IL-12, causes a shift to a more aGvHDlike phenotype (Via et al., 1994). Importantly, the choice of parental donor determines the phenotypic outcome. For example, if C57/Bl6 is the T-cell donor for a B6D2F1 recipient, an aGvHD phenotype results. This might be due to naturally high proportions of cytotoxic CD8+ T cells in the C57/Bl6 mouse strain (Via et al., 1987). Unlike models of aGvHD, which depend primarily on donor T cells, pathology in autoantibody models of cGvHD depends on recipient B cells (Morris et al., 1990a). Specifically, the recipient B cells interact with donor T cells (Morris et al., 1990b) via cell-surface MHC and are also stimulated by cytokines, including IL-4 and IL10 (De Wit et al., 1993; Durie et al., 1994; Via et al., 1996a; Kim et al., 2008). Recipient B cells act as potent APCs that stimulate donor T cells, perpetuating the cycle that leads to autoantibody production. Mouse autoantibody cGvHD models are similar to cGvHD that occurs in humans in that they also involve the production of autoantibodies, although there are a number of distinctions. First, 325 326 Recipient strain C57/Bl6 (H2b) BALB/c Rag2–/– (H2d ) LP/J (H2b) B10.D2 (H2d) None 900-1100 cGy 700-900 cGy Conditioning regimen MHC I mismatched MHC II mismatched 650 cGy No conditioning None or 900 cGy BALB/c (H2d) (C57/Bl6 B6.CH2bm1)F1 (H2b background with mutation at MHC I) (C57/Bl6 B6.CH2bm12)F1 (H2b background with mutation at MHC II) DBA/2 (H2d) C57/Bl6 (H2b) C57/Bl6 (H2b) C3H/HeN (H2k) Mismatched for MHC I, MHC II and miHAs BM-derived CD4+ and CD8+ CD4+ CD8+ – CD4+ CD4+ CD4+ Abbreviations as for Table 1, plus: B6, C57/Bl6; B6D2F1, (C57/Bl6 x DBA/2)F1; ND, not determined. Bl6-H2-Abl –/– (H2b) Thymic dysfunction model 1300 cGy split in two fractions miHA mismatches No conditioning (B6xBALB/c)F1 (H2b/d) C57/Bl6 (H2b) Mismatched for MHC I, MHC II and miHAs Mismatched for MHC I, MHC II and miHAs No conditioning (BALB/cxA)F1 (H2d/a) BALB/c (H2d) Mismatched for MHC I, MHC II and miHAs B6D2F1 (H2b/d) DBA/2 (H2d) ND ND Mismatched for miHAs and MHC H2d matched but miHA mismatched CD4+ Main T-cell type contributing to phenotype miHA mismatched Genetics No conditioning Autoantibody-mediated (lupus-like) models BALB/c (H2d) B10.D2 (H2d) Scleroderma (pro-fibrotic models) Donor strain Table 2. Mouse models of cGVHD 5 106 TCD BM cells No conditioning: 4-5 106 spleen CD4+ T cells, 23 106 BM cells; 2 doses of cells on day 0 and 7 Conditioned: 4-5 106 CD4+ T cells and 1-2 106 TCD BM cells Epidermal atrophy, fat loss, follicular drop out and dermal thickening; bile duct loss and periportal fibrosis in liver; salivary gland fibrosis and atrophy; cytopenias Systemic disease and death by 20-40 days Glomerulonephritis; nonlethal in unirradiated mice; lethal in irradiated mice at 30-60 days 5 107 spleen cells or 108 spleen (2/3) and lymph node (1/3) cells; 2 doses of cells on day 0 and 7 Sakoda et al., 2007 Rolink et al., 1983; Ito et al., 1992; Brown et al., 2002 Rolink et al., 1983; Ito et al., 1992 Zhang et al., 2006 Autoantibodies; skin scleroderma 5 107 whole spleen cells Pals et al., 1985; Vidal et al., 1996 Via et al., 1987; Fujiwara et al., 1991; De Wit et al., 1993; Via et al., 1996a; Slayback et al., 2000; Kim et al., 2006; Kim et al., 2008 Tschetter et al., 2000 Polyarthritis, scleroderma, glomerulonephritis, Sjorgren’s, sclorosing cholangitis Lupus Sjorgren’s (Sialoadenitis); autoantibodies Ruzek et al., 2004 Hamilton and Parkman, 1983; DeClerck et al., 1986 Korngold and Sprent, 1978; Jaffee and Claman, 1983; Claman et al., 1985; Hamilton, 1987; Korngold and Sprent, 1987; McCormick et al., 1999; Levy et al., 2000; Kaplan et al., 2004; Zhou et al., 2007 Reference example(s) Acute progressing to chronic GvHD 6 107 whole spleen cells 8-12 107 spleen cells 1-10 107 Scleroderma; progressive fibrosis of organs; autoantibodies Scleroderma occurring by day 28 20-50 106 spleen cells and 5 107 BM Whole spleen cells (2-5 107) Scleroderma; progressive fibrosis of organs Outcome TCD BM cells and 1 107 whole splenocytes or purified T cells Cells and dose Disease Models & Mechanisms DMM PERSPECTIVE Mouse models of GvHD dmm.biologists.org Mouse models of GvHD nephritis in cGvHD in humans is rare. Second, the repertoire of autoantibodies associated with cGvHD is more diverse in humans than in mice. Finally, lymphadenopathy and splenomegaly do not occur in the human setting. Disease Models & Mechanisms DMM A cGvHD model involving both autoantibodies and scleroderma To more accurately model cGvHD that occurs in humans, attempts have been made to combine the features of autoantibody-mediated and scleroderma models in a single mouse model. For example, transfer of splenocytes from DBA/2 (H2d) mice into sublethally irradiated BALB/c (H2d) recipients results in a lupus-like and sclerodermatous phenotype (Zhang et al., 2006). These mice develop proteinuria with basement membrane immune complex deposits, and skin fibrosis with collagen I deposition; the severity of these disorders depends on the number of donor cells transplanted. In addition, these mice have increased levels of autoantibodies specific for double-stranded DNA. In this case, donor CD4+ T cells are required for the pathology, whereas donor CD8+ T cells do not contribute. Unlike pathology in the parent-toF1 autoantibody-mediated model, which involves recipient B cells, pathology in this model involves donor B cells. Finally, the transfer of TReg cells rescues the cGvHD phenotype, indicating that TReg cells are functional in treating cGvHD, as has been demonstrated in aGvHD models. In summary, this model indicates the importance of donor CD4+ T cells as well as B cells (from both donor and recipient) in the development of cGvHD. However, it does not support a role for the thymus in the pathophysiology of cGvHD, in contrast to the model discussed below. PERSPECTIVE periportal fibrosis in the liver; salivary gland fibrosis and atrophy; and cytopenias (Sakoda et al., 2007). This phenotype developed within 6 weeks of donor cell transfer, and fewer than 20% of animals lived for more than 100 days. Importantly, thymectomized recipient mice abrogated the phenotype, highlighting the role of the thymus in the pathology seen in this model. Experiments involving chimeric mice showed that donor APCs were essential for the development of the cGvHD phenotype in this model: donor APCs that trafficked to the thymus after transplantation were principally responsible for negative selection. In a series of adoptive transfer studies, it was shown that CD4+ T cells isolated from C3H/HeN recipients of MHC-II-deficient bone marrow that develop cGvHD caused aGvHD when transferred to the C57/Bl6 donor strain, but caused cGvHD when transferred to established chimeric mice created from a C57/Bl6 r C3H/HeN transfer. This implies that donor APCs are essential for the development of the cGvHD phenotype not only through their role in negative selection but through peripheral antigen presentation and T-cell stimulation. Although the mouse model described above (H2-Ab1–/– r C3H/HeN) is the first that reproduces multiple clinical features of human cGvHD, it might not be clinically relevant. The model cannot rule out the possibility that additional insult to the thymus caused by GvHD might be the cause of dysfunctional negative selection. It is also difficult to determine the exact kinetics of cGvHD in this model, and whether there is a critical period of thymic damage that is necessary to cause the resulting phenotype. Nonetheless, this model suggests that a crucial component of cGvHD is dysfunctional thymic selection, which may or may not be caused by donor T-cell-induced damage of the recipient thymus as a consequence of aGvHD. Defective thymic function model Given the similarity between the presentation of cGvHD and some autoimmune diseases, as well as the fact that thymectomy of 3day-old mice results in symptoms similar to some aspects of cGvHD (Nishizuka and Sakakura, 1969; Bonomo et al., 1995), it has long been hypothesized that defects in thymic negative selection might have a role in the pathogenesis of cGvHD. The thymus is a crucial target of aGvHD (Fukushi et al., 1990; Krenger et al., 2000b; HauriHohl et al., 2007), and it is known that aGvHD results in destruction of the normal thymic architecture (Ghayur et al., 1988), especially Hassall’s corpuscles, which have been shown to be crucial for the development of TReg cells (Watanabe et al., 2005). The presentation of tissue-restricted antigens by thymic APCs and epithelial cells could result in their direct attack by cytotoxic donor T cells. Cytotoxic attack of these cell types, which are crucial for negative selection, could result in impaired central tolerance and subsequent autoreactivity (Krenger and Hollander, 2008; Mathis and Benoist, 2009). A potential role for thymic damage in GvHD was initially proposed by a group that demonstrated dysfunctional negative selection of autoreactive T-cell clones in mice with aGvHD (van den Brink et al., 2000). More recently, a transgenic mouse transplant model was developed: T-cell-depleted bone marrow from MHCII-deficient mice on a C57/Bl6 background [H2-Ab1–/– (H2b)] were transferred to lethally irradiated C3H/HeN (H2k) recipients. Recipient mice developed a phenotype similar to clinical cGvHD, with skin fibrosis characterized by epidermal atrophy, fat loss, follicular drop out and dermal thickening; bile duct loss and Disease Models & Mechanisms Linking acute and chronic GvHD in a mouse model The high incidence of cGvHD that occurs following aGvHD in humans suggests that there is a direct causal link between these two disease states. If the mechanisms underlying this link could be identified, targeted interventions could be used to prevent the development of autoimmune-like responses that characterize cGvHD from the alloresponses that underlie aGvHD. Zhang et al. attempted to link acute and chronic GvHD in a mouse model (Zhang et al., 2007). Donor bone-marrow-derived CD4+ T cells that arose from hematopoietic precursors following transplantation and were selected (educated) in the recipient thymus during the development of aGvHD could mediate a cGvHD phenotype when transferred to secondary allogeneic recipients, resulting in cGvHD affecting the skin, liver and gut. When transferred to syngeneic secondary recipients, the same cells resulted in a lethal aGvHD phenotype, suggesting that appropriate positive selection had taken place in the recipient thymus but that thymic negative selection was absent or dysfunctional. This illustrates a possible link between the cytotoxic CD8+ T-cell responses of aGvHD and the CD4+ T-cell-mediated responses demonstrated in these models of cGvHD. The exact mechanism is unknown, but these data suggest that the thymus-educated donor CD4+ T cells that should be self-tolerant are autoreactive, suggesting that the link between aGvHD and cGvHD is related to the targeting to the thymus by alloreactive T cells (both CD4+ and CD8+) and subsequent elimination or alteration of the function of thymic cellular elements that are essential for appropriate negative 327 Disease Models & Mechanisms DMM PERSPECTIVE selection. The effects of these autoreactive T cells can only be demonstrated by adoptive transfer experiments because of the highly lethal nature of aGvHD models. Another attempt to investigate the link between aGvHD and cGvHD was performed using an F1 donor and F1 recipient [(B10 ⫻ B10.D2)F1 (H2b/d) r (BALB.b ⫻ BALB/c)F1 (H2b/d)] transplant: this resulted in both systemic symptoms of aGvHD and skin manifestations of cGvHD (Kaplan et al., 2004). In this model, MHC was matched between donor and recipient, but miHAs were mismatched. This same group investigated transplantation between multiple strain combinations matched at MHC loci, and demonstrated that certain MHC haplotypes were associated with the development of GvHD involving specific target organs, which they suspected was due to differences in MHC affinity for antigens found in certain target tissues. For example, immunodominant antigens in the skin might be bound with high affinity in only certain MHC strains. This illustrates the important role of MHC in selecting immunodominant antigens, and how this might influence cGvHD. A final example of the link between alloreactive T-cell responses of aGvHD and the autoimmune responses of cGvHD involved the use of immunodeficient mice (Tivol et al., 2005; Chen et al., 2007). In this case, aGvHD was initiated through a C57/Bl6 r BALB/c lethally irradiated MHC-mismatched transplant. After 20 days, recipient animals were sacrificed and their splenic T cells were isolated. Transfer of these isolated T cells into unconditioned immunodeficient (Rag–/–) C57/Bl6 recipients (i.e. the same background as the original donor) resulted in inflammatory infiltration of the colon and liver (Chen et al., 2007). This autoimmune phenotype was dependent on CD4+ T cells (with a Th1 and Th17 cytokine profile) and could be rescued by the infusion of immunosuppressive donor TReg cells. This supports the data of Zhang et al. described above that a CD4+ T cell – derived either centrally (through the thymus) or peripherally (from the initial donor T-cell inoculum) – in the context of aGvHD can stimulate the autoimmune responses observed in cGvHD. In fact, recent evidence suggests that mature donor-derived CD4+ T cells have the ability to cause both alloreactive and autoreactive responses (Zhao et al., 2010). In a series of adoptive transfer experiments using the DBA/2 r BALB/c cGvHD model, Zhao et al. characterized the CD4+ T cells down to single cells, and showed that a specific TCR predominated in mice with cGvHD. These T cells recognized antigen in the presence of either allogeneic or autologous dendritic cells. Considerations when selecting a mouse model of GvHD With the large number of GvHD models now available, it can be difficult to determine which model is appropriate for the research question at hand. We offer in this section a few suggestions. (1) First, decide whether you are interested in studying acute or chronic GvHD. It is of course useful to first establish from published literature which models have been successfully validated for testing a given intervention. (2) Next, determine what mechanism or T-cell population you want to be dominant in your model for testing a specific therapeutic intervention. For example, a drug that targets CD8+ T cells would not be effective in a model in which CD4+ T cells mediate pathology. Conversely, if limiting the degree of cytokine storm 328 Mouse models of GvHD produced from conditioning is important, then miHA-mismatched or TCR-transgenic models would be useful, because conditioning can be reduced or eliminated. (3) Next, consider measurement outcomes such as: severity, coat color and the ability to track specific cell populations. If easy clinical scoring of GvHD severity and survival are the main outcomes measured, then a model that has consistent rapid kinetics and 100% penetrance is desirable. In this case, the C57/Bl6 r BALB/c transplant model could be a good choice. In general, the less the degree of MHC mismatch, the less severe the model. If bioluminescence imaging (BLI) will be used (see Box 2), consider the coat color of the mouse that will be imaged, because imaging is more difficult with mice that have dark coats (which require shaving prior to BLI). If the tracking of donor cells in the recipient is necessary, it is possible to use flow cytometry to track MHC disparity, to track fluorescent donor cells using BLI, or to use congenic mice that have allelic differences at the CD45 locus (e.g. CD45.1 or CD45.2) to track cellular subsets from donor versus recipient when they have a common MHC. (4) It is also necessary to consider more complex issues such as the production of transgenic strains. Sophisticated modeling can allow for elegant mechanistic studies, but if you are going to use a transgenic strain as a donor or recipient, extensive backcrossing to a single background is essential to eliminate significant genetic disparity that could affect results. (5) Finally, it is crucial to personally validate all new models in your lab. Even a basic model (such as Bl6 r BALB/c) and certainly any novel strain combinations require careful validation to investigate the influence of various factors – including preconditioning radiation dose, mouse strain and cleanliness of colonies (which varies between labs) – on, for example, disease incidence, severity and pathologic evidence of GvHD. When working with any mouse model of GvHD, there are a number of caveats to be aware of when interpreting and translating experimental data, which are outlined in Box 3. Measuring the GVL effect in GvHD models The graft-versus-leukemia (GVL) effect, also referred to as graftversus-tumor effect, describes the alloimmune responses of donor immune cells to residual leukemia or tumor, which are indications for which HSCT is frequently performed as a treatment. Although a complete review of models of GVL is beyond the scope of this article [the reader is instead referred to reviews on the mechanisms Box 2. Imaging pathological events during GvHD Bioluminescent imaging (BLI) has become an important tool for tracking transplanted cells in vivo. This technique has been reviewed recently (Negrin and Contag, 2006). Briefly, this technique uses donor mice that are transgenic for a luciferase-GFP (green fluorescent protein) fusion protein, which is under the control of the constitutively active chicken -actin promoter and cytomegalovirus enhancer [L2G85 mice (FVB background, H2q)]. The activity of the luciferase enzyme results in light emission that can be detected in vivo in recipient mice using a specialized device. This technique is sensitive for visualizing 100-1000 cells per region. It has been used to study the in vivo trafficking and proliferation of T cells (Beilhack et al., 2005), TReg cells (Edinger et al., 2003) and other T-cell subsets, including Th17 cells (Iclozan et al., 2009). It can also be used to measure the GVL effect by tracking the expansion or elimination of luciferase-expressing tumor cells over time after HSCT (Nishimura et al., 2008). dmm.biologists.org PERSPECTIVE Mouse models of GvHD Disease Models & Mechanisms DMM Box 3. Caveats and considerations when interpreting findings from mouse models of GvHD and translating them to the clinic Differences in conditioning regimens Pre-transplant conditioning in humans mainly consists of chemotherapy regimens, given with or without radiation, which is delivered at a rate (typically fractionated) that differs from that used in mouse models of GvHD (usually single dose). Conditioning results in tissue damage and a proinflammatory reaction that might differ between mice and humans and therefore influence the GvHD phenotype. In addition, conditioning regimens and the timing of transplantation can impact experimental outcomes (Gendelman et al., 2004; Mabed et al., 2005; Schwarte and Hoffmann, 2005; Mapara et al., 2006). This can be directly related to the residual host-versus-graft (HVG) effect, which can decrease GvHD severity if the host immune system is not adequately suppressed. The HVG reaction is a rare complication after transplant in humans but increases as conditioning regimen intensity is decreased. Genetic and immunological disparities between donor and recipient In humans, HLA typing occurs via high-resolution DNA typing, and most transplants are matched at the allele level with respect to MHC loci, although they usually differ at the level of miHAs that lie outside of the MHC locus. By contrast, many mouse models of GvHD involve mismatches in both MHC antigens and miHAs. There are also key species differences between the mouse and human immune system that can make the direct translation of results difficult. [These have been reviewed elsewhere (Mestas and Hughes, 2004).] Some of these differences include: involution of the thymus in humans but not mice; expression of MHC II by human but not mouse T cells; and differences in the proportions of circulating lymphocytes and their effector mechanisms in the two species. Furthermore, strain-specific disparities in the proportion of lymphocyte subsets (such as CD4+, CD8+ and TReg cells) influence the phenotype between different mouse models of GvHD. Finally, recent reports suggest that MHC-matched mice can carry genetic polymorphisms outside of the MHC locus that can affect the clinical phenotype of GvHD, which might explain observed differences in results in different labs, even when the same strain combinations are used (Cao et al., 2003; Fanning et al., 2009). Source of donor immune cells Material for HSCT in humans is now most commonly derived from mobilized stem cell products and contains donor immune cells from the circulation. The number, origin and type of circulating immune cells can differ depending on the mobilization regimen used. By contrast, immune cells delivered together with transplants in mouse models are usually isolated from spleen or lymph nodes. It is important to consider that immune-cell populations from different sources might have different trafficking capacities and composition (i.e. different proportions of T-cell subsets, dendritic cells and early myeloid progenitors), and therefore have a different influence on the GvHD phenotype. Differences in normal flora Enteric pathogens can play a role in the kinetics and severity of GvHD, which targets the gut in many cases. Microorganisms express molecules that can activate the immune system and therefore can augment the severity of disease (Cooke et al., 1998). In line with this, changes in the environment of the mice, which in turn affect the composition of gut flora, have been shown to affect disease severity (Bennett et al., 1998; Gerbitz et al., 2004; Gorski et al., 2007). Given that the gut flora of a human is markedly different from that of a mouse kept in a pathogen-free facility, this should be taken into account when translating experimental data. Age HSCT is commonly carried out in adult humans, whereas mice used to study GvHD are typically 8- to 14-weeks old (and have a 2-year lifespan). Given that age can influence the efficiency of immune reconstitution after transplant, as well as susceptibility to GvHD (Ordemann et al., 2002), this factor must be taken into account when translating experimental results. [Adapted, with permission, from Socié and Blazar (Socié and Blazar, 2009).] Disease Models & Mechanisms of GVL (Bleakley and Riddell, 2004; Kolb et al., 2004) as well a recent historical perspective on GVL (Kolb, 2008)], it is worth mentioning that separating GvHD from the GVL effect is a long-standing dilemma in transplantation, and is still an area of active investigation. In fact, separating the immunopathological aspects of GvHD from the curative effect of GVL is perhaps the most clinically meaningful endpoint of HSCT. However, this is a complicated issue because T cells mediate both GvHD and GVL. Research in this area has focused on identifying the mechanisms that might differentiate cytotoxic T-cell responses in GvHD from cytotoxic T-cell responses in GVL, as well as identifying accessory cells that could block GvHD while allowing or augmenting GVL. The first evidence for GVL came from mouse transplant models (Barnes and Loutit, 1957; Weiss et al., 1983). Initial studies in the 1950s demonstrated that a mouse leukemia could not be eliminated by irradiation or by irradiation and syngeneic bone marrow, but irradiation plus allogeneic bone marrow prolonged survival of mice and eliminated leukemia from detection. However, transplanted mice developed a wasting syndrome that later became known as GvHD. Later efforts in the early 1980s were better able to define the immunological basis for this GVL effect, and subsequent work defined the importance of T cells and miHAs, as well as of innate immune cells, such as NK cells, on the GVL effect. The models discussed above can also be used to test GVL effects of allogeneic cellular therapies, as well as to test treatments that are designed to prevent GvHD while preserving the GVL effect. It could be argued that any novel therapy designed in these models to target GvHD should be assessed for its effect on GVL because of the clinical implications for translation to humans. To test the GVL effect, a labeled congenic tumor cell line (i.e. genetically compatible with the recipient, such as the A20 cell line for a BALB/c background) or primary tumor cells [such as mouse acute promyelocytic leukemia cells from PML/RAR transgenic mice (Bl6/129 background) (Westervelt et al., 2003)] can be transduced to stably express a fluorescent marker or luciferase for bioluminescent studies. After a lethal injection of tumor cells at the time of allogeneic transplantation, mice that develop aGvHD generally eliminate the genetically compatible tumor, owing to the alloreactive response mounted by donor T cells. The ultimate goal of interventions has been to prevent GvHD while preserving GVL. There have been a number of recent articles demonstrating this (Liang et al., 2008; Nishimura et al., 2008; Zheng et al., 2009). The availability of BLI (Box 2) has improved the capacity to detect residual leukemia after transplantation and is used as a measure of response to treatments, together with survival and GvHD scoring, in these models. Conclusion Mouse models of GvHD are crucial for advancing our understanding of the disease in its acute and chronic forms. As our understanding grows, it is hoped that targeted therapies will be developed to treat GvHD and preserve GVL. Recent advances in treatment that have evolved owing to studies of mouse models of aGvHD include targeting mediators of the cytokine storm, such as TNF and IL-1, with antagonists (Uberti et al., 2005; Alousi et al., 2009; Antin et al., 2002), as well as reducing the intensity of conditioning regimens to avoid excess inflammation. Other novel interventions include the proteasome inhibitor bortezomib (Sun 329 Disease Models & Mechanisms DMM PERSPECTIVE et al., 2004; Sun et al., 2005), and the DNA methyltransferase inhibitors decitabine and azacitidine (Choi et al., 2010). The exact mechanism by which these agents work is not known, but they might involve suppression of alloreactive T-cell responses and/or an increase in TReg cell number or function following transplantation. In addition, cellular therapies, such as depleting T cells from the graft, or infusing TReg cells, have been developed based on studies performed in the mouse. Finally, cGvHD models have been instrumental in developing imatinib – a tyrosine kinase inhibitor that targets PDGFR, which has been implicated in the pathogenesis of fibrosis (McCormick et al., 1999; Daniels et al., 2004; Abdollahi et al., 2005) – and rituximab, an antibody that targets potential autoreactive B cells (Cutler et al., 2006). Areas that warrant further study include the influence of reduced intensity conditioning regimens (Sadeghi et al., 2008), in vivo imaging, and the genomics and epigenomics of GvHD. As discussed, advances in in vivo imaging techniques to monitor trafficking of immune cells have recently been developed and will be crucial for dissecting the temporal and spatial relationships of donor and recipient T cells, B cells and APCs in these models (Panoskaltsis-Mortari et al., 2004). Furthermore, recent advances in proteomics, gene profiling and whole genome sequencing will help to elucidate the genetic basis of GvHD, provide ways to better match donor and recipient to prevent GvHD, and improve our ability to predict the severity and kinetics of the disease (Baron et al., 2007; Hori et al., 2008; Paczesny et al., 2009). Despite the advances made through preclinical modeling of GvHD in mice, many questions remain. Mouse models will continue to allow for the dissection of mechanistic pathways of GvHD through the use of informative genetic knock-out, knockin and knock-down experiments that are only possible in the mouse. Fundamental research using mouse models of GvHD should enhance our understanding of GvHD in humans and result in improved and novel therapeutic approaches to minimize GvHD while optimizing the GVL effect, as well as immune reconstitution, following HSCT. COMPETING INTERESTS The authors have no relevant financial or competing interests to disclose. REFERENCES Abdollahi, A., Li, M., Ping, G., Plathow, C., Domhan, S., Kiessling, F., Lee, L. B., McMahon, G., Grone, H. J., Lipson, K. E. et al. (2005). Inhibition of platelet-derived growth factor signaling attenuates pulmonary fibrosis. J. Exp. Med. 201, 925-935. Albert, M. H., Liu, Y., Anasetti, C. and Yu, X. Z. (2005). Antigen-dependent suppression of alloresponses by Foxp3-induced regulatory T cells in transplantation. Eur. J. Immunol. 35, 2598-2607. Alousi, A. M., Weisdorf, D. J., Logan, B. R., Bolanos-Meade, J., Carter, S., Difronzo, N., Pasquini, M., Goldstein, S. C., Ho, V. T., Hayes-Lattin, B. et al. (2009). Etanercept, mycophenolate, denileukin, or pentostatin plus corticosteroids for acute graft-versus-host disease: a randomized phase 2 trial from the Blood and Marrow Transplant Clinical Trials Network. Blood 114, 511-517. Anderson, B. E., McNiff, J. M., Jain, D., Blazar, B. R., Shlomchik, W. D. and Shlomchik, M. J. (2005). Distinct roles for donor- and host-derived antigenpresenting cells and costimulatory molecules in murine chronic graft-versus-host disease: requirements depend on target organ. Blood 105, 2227-2234. Antin, J. H., Weisdorf, D., Neuberg, D., Nicklow, R., Clouthier, S., Lee, S. J., Alyea, E., McGarigle, C., Blazar, B. R., Sonis, S. et al. (2002). Interleukin-1 blockade does not prevent acute graft-versus-host disease: results of a randomized, double-blind, placebo-controlled trial of interleukin-1 receptor antagonist in allogeneic bone marrow transplantation. Blood 100, 3479-3482. Baker, M. B., Altman, N. H., Podack, E. R. and Levy, R. B. (1996). The role of cellmediated cytotoxicity in acute GVHD after MHC-matched allogeneic bone marrow transplantation in mice. J. Exp. Med. 183, 2645-2656. 330 Mouse models of GvHD Barnden, M. J., Allison, J., Heath, W. R. and Carbone, F. R. (1998). Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol. Cell Biol. 76, 34-40. Barnes, D. W. and Loutit, J. F. (1957). Treatment of murine leukaemia with x-rays and homologous bone marrow. II. Br. J. Haematol. 3, 241-252. Baron, C., Somogyi, R., Greller, L. D., Rineau, V., Wilkinson, P., Cho, C. R., Cameron, M. J., Kelvin, D. J., Chagnon, P., Roy, D. C. et al. (2007). Prediction of graft-versushost disease in humans by donor gene-expression profiling. PLoS Med. 4, e23. Beilhack, A., Schulz, S., Baker, J., Beilhack, G. F., Wieland, C. B., Herman, E. I., Baker, E. M., Cao, Y. A., Contag, C. H. and Negrin, R. S. (2005). In vivo analyses of early events in acute graft-versus-host disease reveal sequential infiltration of T-cell subsets. Blood 106, 1113-1122. Bennett, M., Taylor, P. A., Austin, M., Baker, M. B., Schook, L. B., Rutherford, M., Kumar, V., Podack, E. R., Mohler, K. M., Levy, R. B. et al. (1998). Cytokine and cytotoxic pathways of NK cell rejection of class I-deficient bone marrow grafts: influence of mouse colony environment. Int. Immunol. 10, 785-790. Berger, M., Wettstein, P. J. and Korngold, R. (1994). T cell subsets involved in lethal graft-versus-host disease directed to immunodominant minor histocompatibility antigens. Transplantation 57, 1095-1102. Blaser, B. W., Roychowdhury, S., Kim, D. J., Schwind, N. R., Bhatt, D., Yuan, W., Kusewitt, D. F., Ferketich, A. K., Caligiuri, M. A. and Guimond, M. (2005). Donorderived IL-15 is critical for acute allogeneic graft-versus-host disease. Blood 105, 894901. Blazar, B. R., Carroll, S. F. and Vallera, D. A. (1991). Prevention of murine graft-versushost disease and bone marrow alloengraftment across the major histocompatibility barrier after donor graft preincubation with anti-LFA1 immunotoxin. Blood 78, 30933102. Bleakley, M. and Riddell, S. R. (2004). Molecules and mechanisms of the graft-versusleukaemia effect. Nat. Rev. Cancer 4, 371-380. Bonomo, A., Kehn, P. J., Payer, E., Rizzo, L., Cheever, A. W. and Shevach, E. M. (1995). Pathogenesis of post-thymectomy autoimmunity. Role of syngeneic MLRreactive T cells. J. Immunol. 154, 6602-6611. Braun, M. Y., Lowin, B., French, L., Acha-Orbea, H. and Tschopp, J. (1996). Cytotoxic T cells deficient in both functional fas ligand and perforin show residual cytolytic activity yet lose their capacity to induce lethal acute graft-versus-host disease. J. Exp. Med. 183, 657-661. Brok, H. P., Vossen, J. M. and Heidt, P. J. (1998). IFN-gamma-mediated prevention of graft-versus-host disease: pharmacodynamic studies and influence on proliferative capacity of chimeric spleen cells. Bone Marrow Transplant. 22, 1005-1010. Brown, G. R., Lee, E. and Thiele, D. L. (2002). TNF-TNFR2 interactions are critical for the development of intestinal graft-versus-host disease in MHC class II-disparate (C57BL/6J->C57BL/6J x bm12)F1 mice. J. Immunol. 168, 3065-3071. Cao, T. M., Lo, B., Ranheim, E. A., Grumet, F. C. and Shizuru, J. A. (2003). Variable hematopoietic graft rejection and graft-versus-host disease in MHC-matched strains of mice. Proc. Natl. Acad. Sci. USA 100, 11571-11576. Carlson, M. J., West, M. L., Coghill, J. M., Panoskaltsis-Mortari, A., Blazar, B. R. and Serody, J. S. (2009). In vitro-differentiated TH17 cells mediate lethal acute graftversus-host disease with severe cutaneous and pulmonary pathologic manifestations. Blood 113, 1365-1374. Chen, X., Vodanovic-Jankovic, S., Johnson, B., Keller, M., Komorowski, R. and Drobyski, W. R. (2007). Absence of regulatory T-cell control of TH1 and TH17 cells is responsible for the autoimmune-mediated pathology in chronic graft-versus-host disease. Blood 110, 3804-3813. Choi, J., Ritchey, J., Prior, J. L., Holt, M., Shannon, W. D., Deych, E., Piwnica-Worms, D. R. and DiPersio, J. F. (2010). In vivo administration of hypomethylating agents mitigate graft-versus-host disease without sacrificing graft-versus-leukemia. Blood 116, 129-139. Christianson, S. W., Greiner, D. L., Hesselton, R. A., Leif, J. H., Wagar, E. J., Schweitzer, I. B., Rajan, T. V., Gott, B., Roopenian, D. C. and Shultz, L. D. (1997). Enhanced human CD4+ T cell engraftment in beta2-microglobulin-deficient NODscid mice. J. Immunol. 158, 3578-3586. Chu, Y. W. and Gress, R. E. (2008). Murine models of chronic graft-versus-host disease: insights and unresolved issues. Biol. Blood Marrow Transplant. 14, 365-378. Claman, H. N., Jaffee, B. D., Huff, J. C. and Clark, R. A. (1985). Chronic graft-versushost disease as a model for scleroderma. II. Mast cell depletion with deposition of immunoglobulins in the skin and fibrosis. Cell. Immunol. 94, 73-84. Cooke, K. R., Kobzik, L., Martin, T. R., Brewer, J., Delmonte, J., Jr, Crawford, J. M. and Ferrara, J. L. (1996). An experimental model of idiopathic pneumonia syndrome after bone marrow transplantation: I. The roles of minor H antigens and endotoxin. Blood 88, 3230-3239. Cooke, K. R., Hill, G. R., Crawford, J. M., Bungard, D., Brinson, Y. S., Delmonte, J., Jr and Ferrara, J. L. (1998). Tumor necrosis factor- alpha production to dmm.biologists.org Disease Models & Mechanisms DMM Mouse models of GvHD lipopolysaccharide stimulation by donor cells predicts the severity of experimental acute graft-versus-host disease. J. Clin. Invest. 102, 1882-1891. Cutler, C., Miklos, D., Kim, H. T., Treister, N., Woo, S. B., Bienfang, D., Klickstein, L. B., Levin, J., Miller, K., Reynolds, C. et al. (2006). Rituximab for steroid-refractory chronic graft-versus-host disease. Blood 108, 756-762. Damdinsuren, B., Zhang, Y., Khalil, A., Wood, W. H., 3rd, Becker, K. G., Shlomchik, M. J. and Sen, R. (2010). Single round of antigen receptor signaling programs naive B cells to receive T cell help. Immunity 32, 355-366. Daniels, C. E., Wilkes, M. C., Edens, M., Kottom, T. J., Murphy, S. J., Limper, A. H. and Leof, E. B. (2004). Imatinib mesylate inhibits the profibrogenic activity of TGFbeta and prevents bleomycin-mediated lung fibrosis. J. Clin. Invest. 114, 1308-1316. De Wit, D., Van Mechelen, M., Zanin, C., Doutrelepont, J. M., Velu, T., Gerard, C., Abramowicz, D., Scheerlinck, J. P., De Baetselier, P., Urbain, J. et al. (1993). Preferential activation of Th2 cells in chronic graft-versus-host reaction. J. Immunol. 150, 361-366. DeClerck, Y., Draper, V. and Parkman, R. (1986). Clonal analysis of murine graft-vshost disease. II. Leukokines that stimulate fibroblast proliferation and collagen synthesis in graft-vs. host disease. J. Immunol. 136, 3549-3552. Durie, F. H., Aruffo, A., Ledbetter, J., Crassi, K. M., Green, W. R., Fast, L. D. and Noelle, R. J. (1994). Antibody to the ligand of CD40, gp39, blocks the occurrence of the acute and chronic forms of graft-vs-host disease. J. Clin. Invest. 94, 1333-1338. Edinger, M., Hoffmann, P., Ermann, J., Drago, K., Fathman, C. G., Strober, S. and Negrin, R. S. (2003). CD4+CD25+ regulatory T cells preserve graft-versus-tumor activity while inhibiting graft-versus-host disease after bone marrow transplantation. Nat. Med. 9, 1144-1150. Eyrich, M., Burger, G., Marquardt, K., Budach, W., Schilbach, K., Niethammer, D. and Schlegel, P. G. (2005). Sequential expression of adhesion and costimulatory molecules in graft-versus-host disease target organs after murine bone marrow transplantation across minor histocompatibility antigen barriers. Biol. Blood Marrow Transplant. 11, 371-382. Fanning, S. L., Appel, M. Y., Berger, S. A., Korngold, R. and Friedman, T. M. (2009). The immunological impact of genetic drift in the B10.BR congenic inbred mouse strain. J. Immunol. 183, 4261-4272. Ferrara, J. L., Abhyankar, S. and Gilliland, D. G. (1993). Cytokine storm of graftversus-host disease: a critical effector role for interleukin-1. Transplant. Proc. 25, 1216-1217. Ferrara, J. L., Levine, J. E., Reddy, P. and Holler, E. (2009). Graft-versus-host disease. Lancet 373, 1550-1561. Fowler, D. H., Breglio, J., Nagel, G., Eckhaus, M. A. and Gress, R. E. (1996). Allospecific CD8+ Tc1 and Tc2 populations in graft-versus-leukemia effect and graftversus-host disease. J. Immunol. 157, 4811-4821. Fujiwara, K., Sakaguchi, N. and Watanabe, T. (1991). Sialoadenitis in experimental graft-versus-host disease. An animal model of Sjogren’s syndrome. Lab. Invest. 65, 710-718. Fukushi, N., Arase, H., Wang, B., Ogasawara, K., Gotohda, T., Good, R. A. and Onoe, K. (1990). Thymus: a direct target tissue in graft-versus-host reaction after allogeneic bone marrow transplantation that results in abrogation of induction of selftolerance. Proc. Natl. Acad. Sci. USA 87, 6301-6305. Gendelman, M., Hecht, T., Logan, B., Vodanovic-Jankovic, S., Komorowski, R. and Drobyski, W. R. (2004). Host conditioning is a primary determinant in modulating the effect of IL-7 on murine graft-versus-host disease. J. Immunol. 172, 3328-3336. Gerbitz, A., Schultz, M., Wilke, A., Linde, H. J., Scholmerich, J., Andreesen, R. and Holler, E. (2004). Probiotic effects on experimental graft-versus-host disease: let them eat yogurt. Blood 103, 4365-4367. Ghayur, T., Seemayer, T. A., Xenocostas, A. and Lapp, W. S. (1988). Complete sequential regeneration of graft-vs.-host-induced severely dysplastic thymuses. Implications for the pathogenesis of chronic graft-vs.-host disease. Am. J. Pathol. 133, 39-46. Gorski, J., Chen, X., Gendelman, M., Yassai, M., Krueger, A., Tivol, E., Logan, B., Komorowski, R., Vodanovic-Jankovic, S. and Drobyski, W. R. (2007). Homeostatic expansion and repertoire regeneration of donor T cells during graft versus host disease is constrained by the host environment. Blood 109, 5502-5510. Graubert, T. A., DiPersio, J. F., Russell, J. H. and Ley, T. J. (1997). Perforin/granzymedependent and independent mechanisms are both important for the development of graft-versus-host disease after murine bone marrow transplantation. J. Clin. Invest. 100, 904-911. Grubin, C. E., Kovats, S., deRoos, P. and Rudensky, A. Y. (1997). Deficient positive selection of CD4 T cells in mice displaying altered repertoires of MHC class II-bound self-peptides. Immunity 7, 197-208. Hakim, F. T., Sharrow, S. O., Payne, S. and Shearer, G. M. (1991). Repopulation of host lymphohematopoietic systems by donor cells during graft-versus-host reaction in unirradiated adult F1 mice injected with parental lymphocytes. J. Immunol. 146, 2108-2115. Disease Models & Mechanisms PERSPECTIVE Hamilton, B. L. (1987). L3T4-positive T cells participate in the induction of graft-vshost disease in response to minor histocompatibility antigens. J. Immunol. 139, 2511-2515. Hamilton, B. L. and Parkman, R. (1983). Acute and chronic graft-versus-host disease induced by minor histocompatibility antigens in mice. Transplantation 36, 150-155. Hauri-Hohl, M. M., Keller, M. P., Gill, J., Hafen, K., Pachlatko, E., Boulay, T., Peter, A., Hollander, G. A. and Krenger, W. (2007). Donor T-cell alloreactivity against host thymic epithelium limits T-cell development after bone marrow transplantation. Blood 109, 4080-4088. Hildebrandt, G. C., Olkiewicz, K. M., Corrion, L. A., Chang, Y., Clouthier, S. G., Liu, C. and Cooke, K. R. (2004). Donor-derived TNF-alpha regulates pulmonary chemokine expression and the development of idiopathic pneumonia syndrome after allogeneic bone marrow transplantation. Blood 104, 586-593. Hill, G. R., Crawford, J. M., Cooke, K. R., Brinson, Y. S., Pan, L. and Ferrara, J. L. (1997). Total body irradiation and acute graft-versus-host disease: the role of gastrointestinal damage and inflammatory cytokines. Blood 90, 3204-3213. Hoffmann-Fezer, G., Gall, C., Zengerle, U., Kranz, B. and Thierfelder, S. (1993). Immunohistology and immunocytology of human T-cell chimerism and graft-versushost disease in SCID mice. Blood 81, 3440-3448. Hori, T., Naishiro, Y., Sohma, H., Suzuki, N., Hatakeyama, N., Yamamoto, M., Sonoda, T., Mizue, Y., Imai, K., Tsutsumi, H. et al. (2008). CCL8 is a potential molecular candidate for the diagnosis of graft-versus-host disease. Blood 111, 44034412. Howell, C. D., Yoder, T., Claman, H. N. and Vierling, J. M. (1989). Hepatic homing of mononuclear inflammatory cells isolated during murine chronic graft-vs-host disease. J. Immunol. 143, 476-483. Iclozan, C., Yu, Y., Liu, C., Liang, Y., Yi, T., Anasetti, C. and Yu, X. Z. (2009). T helper17 cells are sufficient but not necessary to induce acute graft-versus-host disease. Biol. Blood Marrow Transplant. 16, 170-178. Ito, R., Katano, I., Kawai, K., Hirata, H., Ogura, T., Kamisako, T., Eto, T. and Ito, M. (2009). Highly sensitive model for xenogenic GVHD using severe immunodeficient NOG mice. Transplantation 87, 1654-1658. Ito, S., Ueno, M., Nishi, S., Arakawa, M., Ikarashi, Y., Saitoh, T. and Fujiwara, M. (1992). Histological characteristics of lupus nephritis in F1 mice with chronic graftversus-host reaction across MHC class II difference. Autoimmunity 12, 79-87. Jaffee, B. D. and Claman, H. N. (1983). Chronic graft-versus-host disease (GVHD) as a model for scleroderma. I. Description of model systems. Cell. Immunol. 77, 1-12. Jiang, Z., Podack, E. and Levy, R. B. (2001). Major histocompatibility complexmismatched allogeneic bone marrow transplantation using perforin and/or Fas ligand double-defective CD4(+) donor T cells: involvement of cytotoxic function by donor lymphocytes prior to graft-versus-host disease pathogenesis. Blood 98, 390397. Kanamaru, A., Okamoto, T., Matsuda, K., Hara, H. and Nagai, K. (1984). Elevation of erythroid colony-stimulating activity in the serum of mice with graft-versus-host disease. Exp. Hematol. 12, 763-767. Kaplan, D. H., Anderson, B. E., McNiff, J. M., Jain, D., Shlomchik, M. J. and Shlomchik, W. D. (2004). Target antigens determine graft-versus-host disease phenotype. J. Immunol. 173, 5467-5475. Kappel, L. W., Goldberg, G. L., King, C. G., Suh, D. Y., Smith, O. M., Ligh, C., Holland, A. M., Grubin, J., Mark, N. M., Liu, C. et al. (2009). IL-17 contributes to CD4mediated graft-versus-host disease. Blood 113, 945-952. Kim, J., Kim, H. J., Choi, W. S., Nam, S. H., Cho, H. R. and Kwon, B. (2006). Maintenance of CD8+ T-cell anergy by CD4+CD25+ regulatory T cells in chronic graft-versus-host disease. Exp. Mol. Med. 38, 494-501. Kim, J., Park, K., Kim, H. J., Kim, H. A., Jung, D., Choi, H. J., Choi, S. Y., Seo, K. W., Cho, H. R. and Kwon, B. (2008). Breaking of CD8+ T cell tolerance through in vivo ligation of CD40 results in inhibition of chronic graft-versus-host disease and complete donor cell engraftment. J. Immunol. 181, 7380-7389. Kim, Y. M., Sachs, T., Asavaroengchai, W., Bronson, R. and Sykes, M. (2003). Graftversus-host disease can be separated from graft-versus-lymphoma effects by control of lymphocyte trafficking with FTY720. J. Clin. Invest. 111, 659-669. King, M. A., Covassin, L., Brehm, M. A., Racki, W., Pearson, T., Leif, J., Laning, J., Fodor, W., Foreman, O., Burzenski, L. et al. (2009). Human peripheral blood leucocyte non-obese diabetic-severe combined immunodeficiency interleukin-2 receptor gamma chain gene mouse model of xenogeneic graft-versus-host-like disease and the role of host major histocompatibility complex. Clin. Exp. Immunol. 157, 104-118. Kisielow, P., Bluthmann, H., Staerz, U. D., Steinmetz, M. and von Boehmer, H. (1988). Tolerance in T-cell-receptor transgenic mice involves deletion of nonmature CD4+8+ thymocytes. Nature 333, 742-746. Kolb, H. J. (2008). Graft-versus-leukemia effects of transplantation and donor lymphocytes. Blood 112, 4371-4383. Kolb, H. J., Schmid, C., Barrett, A. J. and Schendel, D. J. (2004). Graft-versus-leukemia reactions in allogeneic chimeras. Blood 103, 767-776. 331 Disease Models & Mechanisms DMM PERSPECTIVE Korngold, R. (1992). Lethal graft-versus-host disease in mice directed to multiple minor histocompatibility antigens: features of CD8+ and CD4+ T cell responses. Bone Marrow Transplant. 9, 355-364. Korngold, R. and Sprent, J. (1978). Lethal graft-versus-host disease after bone marrow transplantation across minor histocompatibility barriers in mice. Prevention by removing mature T cells from marrow. J. Exp. Med. 148, 1687-1698. Korngold, R. and Sprent, J. (1987). Variable capacity of L3T4+ T cells to cause lethal graft-versus-host disease across minor histocompatibility barriers in mice. J. Exp. Med. 165, 1552-1564. Krenger, W. and Hollander, G. A. (2008). The immunopathology of thymic GVHD. Semin. Immunopathol. 30, 439-456. Krenger, W., Rossi, S. and Hollander, G. A. (2000a). Apoptosis of thymocytes during acute graft-versus-host disease is independent of glucocorticoids. Transplantation 69, 2190-2193. Krenger, W., Rossi, S., Piali, L. and Hollander, G. A. (2000b). Thymic atrophy in murine acute graft-versus-host disease is effected by impaired cell cycle progression of host pro-T and pre-T cells. Blood 96, 347-354. Lee, S. J. (2010). Have we made progress in the management of chronic graft-vs-host disease? Best Pract. Res Clin. Haematol. 23, 529-535. Levi-Schaffer, F. and Weg, V. B. (1997). Mast cells, eosinophils and fibrosis. Clin. Exp. Allergy 27 Suppl. 1, 64-70. Levy, S., Nagler, A., Okon, S. and Marmary, Y. (2000). Parotid salivary gland dysfunction in chronic graft-versus-host disease (cGVHD): a longitudinal study in a mouse model. Bone Marrow Transplant. 25, 1073-1078. Liang, Y., Liu, C., Djeu, J. Y., Zhong, B., Peters, T., Scharffetter-Kochanek, K., Anasetti, C. and Yu, X. Z. (2008). Beta2 integrins separate graft-versus-host disease and graft-versus-leukemia effects. Blood 111, 954-962. Lu, Y. and Waller, E. K. (2009). Dichotomous role of interferon-gamma in allogeneic bone marrow transplant. Biol. Blood Marrow Transplant. 15, 1347-1353. Lucas, P. J., Shearer, G. M., Neudorf, S. and Gress, R. E. (1990). The human antimurine xenogeneic cytotoxic response. I. Dependence on responder antigenpresenting cells. J. Immunol. 144, 4548-4554. Mabed, M., Maroof, S., Zalta, K. and El-Awadee, M. (2005). Delayed or delayed sequential bone marrow transplantation: relevance for acute graft-versus-host disease prevention after major H2 incompatible transplantation. Bone Marrow Transplant. 35, 803-806. Maeda, Y., Levy, R. B., Reddy, P., Liu, C., Clouthier, S. G., Teshima, T. and Ferrara, J. L. (2005). Both perforin and Fas ligand are required for the regulation of alloreactive CD8+ T cells during acute graft-versus-host disease. Blood 105, 2023-2027. Mapara, M. Y., Leng, C., Kim, Y. M., Bronson, R., Lokshin, A., Luster, A. and Sykes, M. (2006). Expression of chemokines in GVHD target organs is influenced by conditioning and genetic factors and amplified by GVHR. Biol. Blood Marrow Transplant. 12, 623-634. Mathis, D. and Benoist, C. (2009). Aire. Annu. Rev. Immunol. 27, 287-312. McCormick, L. L., Zhang, Y., Tootell, E. and Gilliam, A. C. (1999). Anti-TGF-beta treatment prevents skin and lung fibrosis in murine sclerodermatous graft-versushost disease: a model for human scleroderma. J. Immunol. 163, 5693-5699. Mestas, J. and Hughes, C. C. (2004). Of mice and not men: differences between mouse and human immunology. J. Immunol. 172, 2731-2738. Morris, S. C., Cheek, R. L., Cohen, P. L. and Eisenberg, R. A. (1990a). Allotype-specific immunoregulation of autoantibody production by host B cells in chronic graftversus host disease. J. Immunol. 144, 916-922. Morris, S. C., Cheek, R. L., Cohen, P. L. and Eisenberg, R. A. (1990b). Autoantibodies in chronic graft versus host result from cognate T-B interactions. J. Exp. Med. 171, 503-517. Mosier, D. E., Gulizia, R. J., Baird, S. M. and Wilson, D. B. (1988). Transfer of a functional human immune system to mice with severe combined immunodeficiency. Nature 335, 256-259. Murphy, K. M., Heimberger, A. B. and Loh, D. Y. (1990). Induction by antigen of intrathymic apoptosis of CD4+CD8+TCRlo thymocytes in vivo. Science 250, 17201723. Negrin, R. S. and Contag, C. H. (2006). In vivo imaging using bioluminescence: a tool for probing graft-versus-host disease. Nat. Rev. Immunol. 6, 484-490. Nervi, B., Rettig, M. P., Ritchey, J. K., Wang, H. L., Bauer, G., Walker, J., Bonyhadi, M. L., Berenson, R. J., Prior, J. L., Piwnica-Worms, D. et al. (2007). Factors affecting human T cell engraftment, trafficking, and associated xenogeneic graft-vs-host disease in NOD/SCID beta2mnull mice. Exp. Hematol. 35, 1823-1838. Nestel, F. P., Price, K. S., Seemayer, T. A. and Lapp, W. S. (1992). Macrophage priming and lipopolysaccharide-triggered release of tumor necrosis factor alpha during graftversus-host disease. J. Exp. Med. 175, 405-413. Niculescu, F., Niculescu, T., Nguyen, P., Puliaev, R., Papadimitriou, J. C., Gaspari, A., Rus, H. and Via, C. S. (2005). Both apoptosis and complement membrane attack complex deposition are major features of murine acute graft-vs.-host disease. Exp. Mol. Pathol. 79, 136-145. 332 Mouse models of GvHD Nikolic, B., Lee, S., Bronson, R. T., Grusby, M. J. and Sykes, M. (2000). Th1 and Th2 mediate acute graft-versus-host disease, each with distinct end-organ targets. J. Clin. Invest. 105, 1289-1298. Nishimura, R., Baker, J., Beilhack, A., Zeiser, R., Olson, J. A., Sega, E. I., Karimi, M. and Negrin, R. S. (2008). In vivo trafficking and survival of cytokine-induced killer cells resulting in minimal GVHD with retention of antitumor activity. Blood 112, 2563-2574. Nishizuka, Y. and Sakakura, T. (1969). Thymus and reproduction: sex-linked dysgenesia of the gonad after neonatal thymectomy in mice. Science 166, 753-755. Ordemann, R., Hutchinson, R., Friedman, J., Burakoff, S. J., Reddy, P., Duffner, U., Braun, T. M., Liu, C., Teshima, T. and Ferrara, J. L. (2002). Enhanced allostimulatory activity of host antigen-presenting cells in old mice intensifies acute graft-versushost disease. J. Clin. Invest. 109, 1249-1256. Paczesny, S., Krijanovski, O. I., Braun, T. M., Choi, S. W., Clouthier, S. G., Kuick, R., Misek, D. E., Cooke, K. R., Kitko, C. L., Weyand, A. et al. (2009). A biomarker panel for acute graft-versus-host disease. Blood 113, 273-278. Pals, S. T., Radaszkiewicz, T., Roozendaal, L. and Gleichmann, E. (1985). Chronic progressive polyarthritis and other symptoms of collagen vascular disease induced by graft-vs-host reaction. J. Immunol. 134, 1475-1482. Panoskaltsis-Mortari, A., Lacey, D. L., Vallera, D. A. and Blazar, B. R. (1998). Keratinocyte growth factor administered before conditioning ameliorates graftversus-host disease after allogeneic bone marrow transplantation in mice. Blood 92, 3960-3967. Panoskaltsis-Mortari, A., Price, A., Hermanson, J. R., Taras, E., Lees, C., Serody, J. S. and Blazar, B. R. (2004). In vivo imaging of graft-versus-host-disease in mice. Blood 103, 3590-3598. Pickel, K. and Hoffmann, M. K. (1977). Suppressor T cells arising in mice undergoing a graft-vs-host response. J. Immunol. 118, 653-656. Ringden, O., Karlsson, H., Olsson, R., Omazic, B. and Uhlin, M. (2009). The allogeneic graft-versus-cancer effect. Br. J. Haematol. 147, 614-633. Rolink, A. G. and Gleichmann, E. (1983). Allosuppressor- and allohelper-T cells in acute and chronic graft-vs.-host (GVH) disease. III. Different Lyt subsets of donor T cells induce different pathological syndromes. J. Exp. Med. 158, 546-558. Rolink, A. G., Pals, S. T. and Gleichmann, E. (1983). Allosuppressor and allohelper T cells in acute and chronic graft-vs.-host disease. II. F1 recipients carrying mutations at H-2K and/or I-A. J. Exp. Med. 157, 755-771. Ruzek, M. C., Jha, S., Ledbetter, S., Richards, S. M. and Garman, R. D. (2004). A modified model of graft-versus-host-induced systemic sclerosis (scleroderma) exhibits all major aspects of the human disease. Arthritis Rheum. 50, 1319-1331. Sadeghi, B., Aghdami, N., Hassan, Z., Forouzanfar, M., Rozell, B., Abedi-Valugerdi, M. and Hassan, M. (2008). GVHD after chemotherapy conditioning in allogeneic transplanted mice. Bone Marrow Transplant. 42, 807-818. Sakaguchi, S., Ono, M., Setoguchi, R., Yagi, H., Hori, S., Fehervari, Z., Shimizu, J., Takahashi, T. and Nomura, T. (2006). Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol. Rev. 212, 8-27. Sakoda, Y., Hashimoto, D., Asakura, S., Takeuchi, K., Harada, M., Tanimoto, M. and Teshima, T. (2007). Donor-derived thymic-dependent T cells cause chronic graftversus-host disease. Blood 109, 1756-1764. Schwarte, S. and Hoffmann, M. W. (2005). Influence of radiation protocols on graftvs-host disease incidence after bone-marrow transplantation in experimental models. Methods Mol. Med. 109, 445-458. Schwarte, S., Bremer, M., Fruehauf, J., Sorge, Y., Skubich, S. and Hoffmann, M. W. (2007). Radiation protocols determine acute graft-versus-host disease incidence after allogeneic bone marrow transplantation in murine models. Int. J. Radiat. Biol. 83, 625-636. Sha, W. C., Nelson, C. A., Newberry, R. D., Kranz, D. M., Russell, J. H. and Loh, D. Y. (1988). Positive and negative selection of an antigen receptor on T cells in transgenic mice. Nature 336, 73-76. Shimabukuro-Vornhagen, A., Hallek, M. J., Storb, R. F. and von Bergwelt-Baildon, M. S. (2009). The role of B Cells in the pathogenesis of graft-versus-host disease. Blood 114, 4919-4927. Shlomchik, W. D., Couzens, M. S., Tang, C. B., McNiff, J., Robert, M. E., Liu, J., Shlomchik, M. J. and Emerson, S. G. (1999). Prevention of graft versus host disease by inactivation of host antigen-presenting cells. Science 285, 412-415. Skert, C., Patriarca, F., Sperotto, A., Cerno, M., Fili, C., Zaja, F., Stocchi, R., Geromin, A., Damiani, D. and Fanin, R. (2006). Sclerodermatous chronic graft-versus-host disease after allogeneic hematopoietic stem cell transplantation: incidence, predictors and outcome. Haematologica 91, 258-261. Slayback, D. L., Dobkins, J. A., Harper, J. M. and Allen, R. D. (2000). Genetic factors influencing the development of chronic graft-versus-host disease in a murine model. Bone Marrow Transplant. 26, 931-938. Socié, G. and Blazar, B. R. (2009). Acute graft-versus-host disease; from the bench to the bedside. Blood 114, 4327-4336. dmm.biologists.org Disease Models & Mechanisms DMM Mouse models of GvHD Sprent, J., Schaefer, M., Lo, D. and Korngold, R. (1986). Properties of purified T cell subsets. II. In vivo responses to class I vs. class II H-2 differences. J. Exp. Med. 163, 998-1011. Sprent, J., Schaefer, M., Gao, E. K. and Korngold, R. (1988). Role of T cell subsets in lethal graft-versus-host disease (GVHD) directed to class I versus class II H-2 differences. I. L3T4+ cells can either augment or retard GVHD elicited by Lyt-2+ cells in class I different hosts. J. Exp. Med. 167, 556-569. Sprent, J., Schaefer, M. and Korngold, R. (1990). Role of T cell subsets in lethal graftversus-host disease (GVHD) directed to class I versus class II H-2 differences. II. Protective effects of L3T4+ cells in anti-class II GVHD. J. Immunol. 144, 2946-2954. Sun, K., Welniak, L. A., Panoskaltsis-Mortari, A., O’Shaughnessy, M. J., Liu, H., Barao, I., Riordan, W., Sitcheran, R., Wysocki, C., Serody, J. S. et al. (2004). Inhibition of acute graft-versus-host disease with retention of graft-versus-tumor effects by the proteasome inhibitor bortezomib. Proc. Natl. Acad. Sci. USA 101, 81208125. Sun, K., Wilkins, D. E., Anver, M. R., Sayers, T. J., Panoskaltsis-Mortari, A., Blazar, B. R., Welniak, L. A. and Murphy, W. J. (2005). Differential effects of proteasome inhibition by bortezomib on murine acute graft-versus-host disease (GVHD): delayed administration of bortezomib results in increased GVHD-dependent gastrointestinal toxicity. Blood 106, 3293-3299. Svegliati, S., Olivieri, A., Campelli, N., Luchetti, M., Poloni, A., Trappolini, S., Moroncini, G., Bacigalupo, A., Leoni, P., Avvedimento, E. V. et al. (2007). Stimulatory autoantibodies to PDGF receptor in patients with extensive chronic graft-versus-host disease. Blood 110, 237-241. Tawara, I., Maeda, Y., Sun, Y., Lowler, K. P., Liu, C., Toubai, T., McKenzie, A. N. and Reddy, P. (2008). Combined Th2 cytokine deficiency in donor T cells aggravates experimental acute graft-vs-host disease. Exp. Hematol. 36, 988-996. Teshima, T., Ordemann, R., Reddy, P., Gagin, S., Liu, C., Cooke, K. R. and Ferrara, J. L. (2002). Acute graft-versus-host disease does not require alloantigen expression on host epithelium. Nat. Med. 8, 575-581. Tivol, E., Komorowski, R. and Drobyski, W. R. (2005). Emergent autoimmunity in graft-versus-host disease. Blood 105, 4885-4891. Tschetter, J. R., Mozes, E. and Shearer, G. M. (2000). Progression from acute to chronic disease in a murine parent-into-F1 model of graft-versus-host disease. J. Immunol. 165, 5987-5994. Uberti, J. P., Ayash, L., Ratanatharathorn, V., Silver, S., Reynolds, C., Becker, M., Reddy, P., Cooke, K. R., Yanik, G., Whitfield, J. et al. (2005). Pilot trial on the use of etanercept and methylprednisolone as primary treatment for acute graft-versus-host disease. Biol. Blood Marrow Transplant. 11, 680-687. Vallera, D. A., Soderling, C. C., Carlson, G. J. and Kersey, J. H. (1981). Bone marrow transplantation across major histocompatibility barriers in mice. Effect of elimination of T cells from donor grafts by treatment with monoclonal Thy-1.2 plus complement or antibody alone. Transplantation 31, 218-222. van den Brink, M. R., Moore, E., Ferrara, J. L. and Burakoff, S. J. (2000). Graft-versushost-disease-associated thymic damage results in the appearance of T cell clones with anti-host reactivity. Transplantation 69, 446-449. van Leeuwen, L., Guiffre, A., Atkinson, K., Rainer, S. P. and Sewell, W. A. (2002). A two-phase pathogenesis of graft-versus-host disease in mice. Bone Marrow Transplant. 29, 151-158. van Rijn, R. S., Simonetti, E. R., Hagenbeek, A., Hogenes, M. C., de Weger, R. A., Canninga-van Dijk, M. R., Weijer, K., Spits, H., Storm, G., van Bloois, L. et al. (2003). A new xenograft model for graft-versus-host disease by intravenous transfer of human peripheral blood mononuclear cells in RAG2-/- gammac-/- double-mutant mice. Blood 102, 2522-2531. Via, C. S., Sharrow, S. O. and Shearer, G. M. (1987). Role of cytotoxic T lymphocytes in the prevention of lupus-like disease occurring in a murine model of graft-vs-host disease. J. Immunol. 139, 1840-1849. Via, C. S., Rus, V., Gately, M. K. and Finkelman, F. D. (1994). IL-12 stimulates the development of acute graft-versus-host disease in mice that normally would Disease Models & Mechanisms PERSPECTIVE develop chronic, autoimmune graft-versus-host disease. J. Immunol. 153, 40404047. Via, C. S., Rus, V., Nguyen, P., Linsley, P. and Gause, W. C. (1996a). Differential effect of CTLA4Ig on murine graft-versus-host disease (GVHD) development: CTLA4Ig prevents both acute and chronic GVHD development but reverses only chronic GVHD. J. Immunol. 157, 4258-4267. Via, C. S., Nguyen, P., Shustov, A., Drappa, J. and Elkon, K. B. (1996b). A major role for the Fas pathway in acute graft-versus-host disease. J. Immunol. 157, 5387-5393. Vidal, S., Labrador, M., Rodriguez-Sanchez, J. L. and Gelpi, C. (1996). The role of BALB/c donor CD8+ lymphocytes in graft-versus-host disease in (BALB/c x A/J)F1 (CAF1) mice. J. Immunol. 156, 997-1005. Wang, X., Roopenian, D., Martone, C., Li, N., Li, H. and Shlomchik, W. D. (2009). Mechanisms of cross-presentation in graft-vs-host disease. Blood 114, (ASH Annual Meeting Abstracts), Abstract 687. Watanabe, N., Wang, Y. H., Lee, H. K., Ito, T., Cao, W. and Liu, Y. J. (2005). Hassall’s corpuscles instruct dendritic cells to induce CD4+CD25+ regulatory T cells in human thymus. Nature 436, 1181-1185. Weiss, L., Morecki, S., Vitetta, E. S. and Slavin, S. (1983). Suppression and elimination of BCL1 leukemia by allogeneic bone marrow transplantation. J. Immunol. 130, 24522455. Westervelt, P., Lane, A. A., Pollock, J. L., Oldfather, K., Holt, M. S., Zimonjic, D. B., Popescu, N. C., DiPersio, J. F. and Ley, T. J. (2003). High-penetrance mouse model of acute promyelocytic leukemia with very low levels of PML-RARalpha expression. Blood 102, 1857-1865. Wilkinson, K. N., Anderson, B., McNiff, J., Demetris, A. J., Shlomchik, W. D. and Shlomchik, M. (2009). A new TCR transgenic model of GVHD reveals that, independent of repertoire, effector memory T cells are severely limited, and central memory T cells somewhat limited, in their ability to cause GVHD. Blood 114 (ASH Annual Meeting Abstracts), Abstract 233. Wynn, T. A. (2004). Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat. Rev. Immunol. 4, 583-594. Wysocki, C. A., Panoskaltsis-Mortari, A., Blazar, B. R. and Serody, J. S. (2005). Leukocyte migration and graft-versus-host disease. Blood 105, 4191-4199. Yi, T., Zhao, D., Lin, C. L., Zhang, C., Chen, Y., Todorov, I., LeBon, T., Kandeel, F., Forman, S. and Zeng, D. (2008). Absence of donor Th17 leads to augmented Th1 differentiation and exacerbated acute graft-versus-host disease. Blood 112, 21012110. Yu, X. Z., Albert, M. H. and Anasetti, C. (2006). Alloantigen affinity and CD4 help determine severity of graft-versus-host disease mediated by CD8 donor T cells. J. Immunol. 176, 3383-3390. Zhang, C., Todorov, I., Zhang, Z., Liu, Y., Kandeel, F., Forman, S., Strober, S. and Zeng, D. (2006). Donor CD4+ T and B cells in transplants induce chronic graftversus-host disease with autoimmune manifestations. Blood 107, 2993-3001. Zhang, Y., Joe, G., Hexner, E., Zhu, J. and Emerson, S. G. (2005). Alloreactive memory T cells are responsible for the persistence of graft-versus-host disease. J. Immunol. 174, 3051-3058. Zhang, Y., Hexner, E., Frank, D. and Emerson, S. G. (2007). CD4+ T cells generated de novo from donor hemopoietic stem cells mediate the evolution from acute to chronic graft-versus-host disease. J. Immunol. 179, 3305-3314. Zhao, D., Young, J. S., Chen, Y. H., Shen, E., Yi, T., Todorov, I., Chu, P. G., Forman, S. J. and Zeng, D. (2010). Alloimmune response results in expansion of autoreactive donor CD4+ T cells in transplants that can mediate chronic graft-versus-host disease. J. Immunol. 186, 856-868. Zheng, H., Matte-Martone, C., Jain, D., McNiff, J. and Shlomchik, W. D. (2009). Central memory CD8+ T cells induce graft-versus-host disease and mediate graftversus-leukemia. J. Immunol. 182, 5938-5948. Zhou, L., Askew, D., Wu, C. and Gilliam, A. C. (2007). Cutaneous gene expression by DNA microarray in murine sclerodermatous graft-versus-host disease, a model for human scleroderma. J. Invest. Dermatol. 127, 281-292. 333