Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Schistosoma mansoni wikipedia , lookup
Hepatitis B wikipedia , lookup
Herpes simplex virus wikipedia , lookup
Cross-species transmission wikipedia , lookup
Sexually transmitted infection wikipedia , lookup
Antiviral drug wikipedia , lookup
Epidemiology of HIV/AIDS wikipedia , lookup
Diagnosis of HIV/AIDS wikipedia , lookup
Microbicides for sexually transmitted diseases wikipedia , lookup
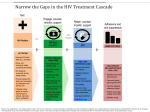
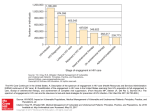
IAS Workshop 2 July 2013 Combining –omics to study the host and the virus Jacques Fellay School of Life Sciences École Polytechnique Fédérale de Lausanne - EPFL Lausanne, Switzerland Allele frequency of variant <<<<<1% >5% Sequencing studies +++ How to look for associations with DNA variants? ++ Clinical impact Genome-wide association studies + HIV host genetic studies: clinical phenotypes Viral control disease progression Resistance acquisition Exposure Infection Science 2007 Aug 17;317(5840):944-7 Science 2010 Dec 10;330(6010):1551-7 Science 1996 Sep 27;273(5283):1856-62 Where do we go from here? 1. More common variants? Meta-analysis of GWAS data 2. Rare functional variants? Sequencing 3. Host impact on viral sequence? “Genome-to-genome” interaction analysis More common variants? International Collaboration for the Genomics of HIV Objective: combine existing GWAS data from HIV+ cohorts to conduct joint analyses: - of viral control and/or disease progression - of HIV susceptibility: After QC and imputation, comparison between 6300 HIV infected cases and 7300 population controls over 5x106 variants B*57:01 B*27:05 Frailty bias Due to their shorter survival time, patients with rapid disease progression are underrepresented in “chronic” cohorts, while individuals with prolonged disease-free survival times are more likely to be included Analysis restricted to patients with known date of infection rs4418214 p=0.01 International Collaboration for the Genomics of HIV HIV acquisition: no significant associations (after accounting for survivor bias), with the exception of CCR5Δ32 homozygosity: p=3E13. No replication of all other previously reported associations (N=22) McLaren et al., PLoS Pathogens, in press HIV control: analyses are ongoing Rare functional variants? Polymorphisms of strong effect are kept at low frequency by evolutionary forces Rare, functional variants are not well represented by GWAS Sequencing has been highly successful for uncovering causes of rare Mendelian diseases Patient sample N=400 Target enrichment Variant calling and frequency estimation www.broadinstitute.org/gatk DNA extraction and quantity normalization Sequencing (paired end reads) Variant annotation CAA GTA AAC ATA GGA CTT CTT CAA GTA AAC ATA GGA CAT CTT snpeff.sourceforge.net DNA pooling and bar-coding Association testing with HIV VL Alignment and base quality recalibration T/C Single variant Gene burden Exome sequencing performance Metric Mean coverage % Covered >5x Call rate GWAS concordance Per sample Total non-ref Non-synonymous Loss of function Ti/Tv Score 73x 94.0% 99.9% 99.0% Score 16,105 8,122 39 3.21 Single variant results (MAF > 1%) MHC signal consistent with GWAS Can be explained by variation in HLA-B (B*57:01) and HLA-C (3’ UTR) Single variant results (MAF > 1%) No single variant associates with spVL after accounting for known signals Burden testing • Gene-based (~20,000 tests) • Set-based siRNA Screens Interacting Proteins Burden testing HIV-specific sets from the literature I HIV dependency factors II HIV/Human PPI by MS III Interferon stimulated genes IV HIV interactome No significant Union set = associations 2,791 Intersection (2 or more) = 292 Restrict analysis to nonsynonymous and loss of function variants Host genomics of HIV disease: Limitations of clinical phenotypes 1. Good phenotypes are hard to get: - Long follow-up of patients - Close collaboration with clinicians - It’s now unethical to observe the natural history of HIV infection 2. Clinical outcomes are quite far from potentially causal gene variants Host genomics Host-pathogen genomics The principle of Genome-to-Genome analysis Escape mutations Genetic variants Host restriction factors leading to viral escape can be uncovered by searching for their imprints on viral genomes HIV-1 “genome-to-genome” study • 1100 study participants • Caucasians infected with subtype B HIV-1 • Paired genetic data: Human: genome-wide genotypes from GWAS HIV-1: full-length consensus sequence 3 sets of genome-wide comparisons Human genetic variation 1 GWAS Viral load 2077 GWAS (1 per variable HIV amino acid present in >20 samples) 1 proteome-wide association study (2077 linear regressions) HIV-1 amino acid variants Human SNPs Viral Load HIV sequence mutations Human SNPs Viral Load HIV sequence mutations SNPs, HLA and CTL epitopes Association of HIV-1 amino acids with VL Human SNPs Viral Load HIV sequence mutations No significant association Changes in VL for amino acid variants associated with rs2395029 / B*57:01 (p<0.001) Conclusions • Using viral variation as an intermediate phenotype can be a sensitive method for detecting host associations • Can be applied to other infectious diseases HIV host genetics – the way forward 1. More samples – Host genetics of infectious disease outcome still lags far behind other complex traits in terms of power 2. More variants – Current technologies still do not provide a complete picture of human genetic variation 3. More phenotypes – Easily measured, intermediate phenotypes can provide a potentially powerful method for detection of important loci Paul McLaren Istvan Bartha Thomas Junier Samira Asgari Ana Bittencourt All ICGH collaborators University of Lausanne Microsoft Research Duke University Genomic Technologies Facility Vital-IT Computing Center Amalio Telenti David Heckerman David Goldstein Keith Harshman Ioannis Xenarios