Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
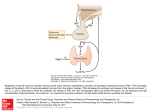
Pharmacology & Therapeutics 121 (2009) 20–28 Contents lists available at ScienceDirect Pharmacology & Therapeutics j o u r n a l h o m e p a g e : w w w. e l s ev i e r. c o m / l o c a t e / p h a r m t h e r a The thyrotropin-releasing hormone (TRH)–immune system homeostatic hypothesis J. Kamath a,⁎, G.G. Yarbrough b, A.J. Prange Jr. c, A. Winokur a a b c University of Connecticut Health Center, Department of Psychiatry, 263 Farmington Avenue, Farmington, CT 06030, United States TRH Therapeutics LLC, 2366 NW Pettygrove Street, Portland, OR 97210, United States University of North Carolina, Department of Psychiatry, 6503 Meadow View Road, Hillsborough, NC 27278, United States a r t i c l e i n f o Keywords: TRH Thyrotropin-releasing hormone Immune cytokine Inflammation Homeostasis Abbreviations: CNS, central nervous system CRH, corticotropin-releasing hormone D2, type 2 deiodinase DMV, dorsal motor nucleus of the vagus HPA, hypothalamic-pituitary–adrenocortical HPT, hypothalamic-pituitary–thyroid IEL, intraepithelial lymphocytes IFN-γ, interferon gamma IL-1, interleukin-1 IL-2, interleukin-2 IL-6, interleukin-6 LPS, lipopolysaccharide NTS, nucleus tractus solitarius PBMC, peripheral blood mononuclear cells PRL, prolactin PVN, paraventricular nuclei SRBC, sheep red blood cells TNF-α, tumor necrosis factor-alpha TRH, thyrotropin-releasing hormone TRH-R, TRH receptor TSH, thyroid stimulating hormone VLM, ventrolateral medulla a b s t r a c t Decades of research have established that the biological functions of thyrotropin-releasing hormone (TRH) extend far beyond its role as a regulator of the hypothalamic-pituitary–thyroid axis. Gary et al. [Gary, K.A., Sevarino, K.A., Yarbrough, G.G., Prange, A.J. Jr., Winokur, A. (2003). The thyrotropin-releasing hormone (TRH) hypothesis of homeostatic regulation: implications for TRH-based therapeutics. J Pharmacol Exp Ther 305(2):410–416.] and Yarbrough et al. [Yarbrough, G.G., Kamath, J., Winokur, A., Prange, A.J. Jr. (2007). Thyrotropin-releasing hormone (TRH) in the neuroaxis: therapeutic effects reflect physiological functions and molecular actions. Med Hypotheses 69(6):1249–1256.] provided a functional framework, predicated on its global homeostatic influences, to conceptualize the numerous interactions of TRH with the central nervous system (CNS) and endocrine system. Herein, we profer a similar analysis to interactions of TRH with the immune system. Autocrine/paracrine cellular signaling motifs of TRH and TRH receptors are expressed in several tissues and organs of the immune system. Consistent with this functional distribution, in vitro and in vivo evidence suggests a critical role for TRH during the developmental stages of the immune system as well as its numerous interactions with the fully developed immune system. Considerable evidence supports a pivotal role for TRH in the pathophysiology of the inflammatory process with specific relevance to the “cytokine-induced sickness behavior” paradigm. These findings, combined with a number of documented clinical actions of TRH strongly support a potential utility of TRH-based therapeutics in select inflammatory disorders. Similar to its global role in behavioral and energy homeostasis a homeostatic role for TRH in its interactions with the immune system is consonant with the large body of available data. Recent advances in the field of immunology provide a significant opportunity for investigation of the TRH-immune system homeostatic hypothesis. Moreover, this hypothesis may provide a foundation for the development of TRH-based therapeutics for certain medical and psychiatric disorders involving immune dysfunction. © 2008 Elsevier Inc. All rights reserved. Contents 1. 2. 3. 4. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . The expression of thyrotropin-releasing hormone and thyrotropin-releasing hormone-receptor in the immune system . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . In vitro evidence for interactions between thyrotropin-releasing hormone and the immune system . . . . . . In vivo evidence for interactions between thyrotropin-releasing hormone and the immune system . . . . . . 4.1. Effects of thyrotropin-releasing hormone on the immune system. . . . . . . . . . . . . . . . . . . 4.2. Effects of cytokines on thyrotropin-releasing hormone systems . . . . . . . . . . . . . . . . . . . 4.3. Role for thyrotropin-releasing hormone in the regulation of immune responses. . . . . . . . . . . . 21 21 22 22 22 23 23 ⁎ Corresponding author. University of Connecticut Health Center, University of Connecticut School of Medicine, Department of Psychiatry, 10 Talcott Notch Road, East Lobby, 3rd Floor, Farmington CT 06030-6415, United States. Tel.: 860 679 6727; fax: 860 679 6781. E-mail address: [email protected] (J. Kamath). 0163-7258/$ – see front matter © 2008 Elsevier Inc. All rights reserved. doi:10.1016/j.pharmthera.2008.09.004 J. Kamath et al. / Pharmacology & Therapeutics 121 (2009) 20–28 5. Connections between thyrotropin-releasing hormone and the immune system . . . . . . . . . . . . . 5.1. Anatomical framework for thyrotropin-releasing hormone–immune system interactions . . . . . 5.2. Experimental evidence for vagus-dependent thyrotropin-releasing hormone–immune system interactions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5.3. Experimental evidence for vagus-independent thyrotropin-releasing hormone–immune system interactions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5.4. Role of thyrotropin-releasing hormone in the pathophysiology of the inflammatory process. . . . 6. Thyrotropin-releasing hormone-based therapeutics in inflammatory disorders . . . . . . . . . . . . . 6.1. Cytokine-induced sickness syndrome as a therapeutic target in inflammatory disorders . . . . . . 6.2. Challenges and advances in the development of thyrotropin-releasing hormone-based therapeutics 7. Future investigations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 8. Conclusions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1. Introduction Ader first used the term “psychoneuroimmunology” in 1980 (Ader, 1980) to describe the increasing body of evidence about interactions between the brain and the immune system. Since then research has demonstrated significant involvement of the endocrine system in brain-immune interactions in both health and disease states. These multi-directional interactions can occur at many stages of development, and they are a continual part of the drive to maintain homeostasis. Defects or deficiencies in one or more of these systems can lead to a specific disorder or aggravate a variety of other disorders. Interactions between the central nervous system (CNS), the endocrine system and the immune system are mediated at multiple levels. These mediators include secreted chemical messengers such as hormones, cytokines, neurotransmitters and neuropeptides acting directly or via the nervous system. Evidence indicates interactions at the level of receptors (e.g., the presence of neuroendocrine peptide receptors on immune cells), at the level of secretory function (e.g., the synthesis and secretion of neuroendocrine peptides by immune cells), and at the level of signal transduction. Interactions of specific endocrine systems (e.g., the hypothalamicpituitary–adrenocortical [HPA] axis) with the CNS and the immune system have been extensively described (Sternberg, 1995; Eskandari & Sternberg, 2002). Hypophysectomized rats and mice exhibit decreased antibody response, decreased lymphocyte proliferation, reduced spleen natural killer cell activity, and prolongation of graft survival (Keller et al., 1988; Nagy & Berczi, 1978). Despite the increasing evidence in individual reports, few reviews have attempted to formulate interactions between the hypothalamic-pituitary–thyroid (HPT) axis, the CNS and the immune system (Pawlikowski et al., 1994; Kruger, 1996). To date, no review has delineated the interactions of one of the critical hormones of the HPT axis, thyrotropin-releasing hormone (TRH), with immune and other systems. In the present report, we first review and discuss some of the pivotal evidence for interactions between TRH, the CNS and the immune system and then propose a functional framework in which to conceptualize the accumulating evidence. We propose a TRH-immune system homeostatic hypothesis. The TRH-immune system homeostatic hypothesis states that TRHmediated mechanisms respond to many elements of the immune system and affect them in ways that tend to maintain or restore homeostasis. Finally, we describe potential implications of this framework for the pathophysiology of certain disorders and provide a rationale for TRH-based therapeutics. The tripeptide thyrotropin-releasing hormone (TRH) is known to control the synthesis and secretion of pituitary thyrotropin (thyroid stimulating hormone, TSH) and prolactin (PRL) (comprehensive review in Nillni & Sevarino, 1999). TRH-secreting neurons are located in the medial portions of the paraventricular nuclei (PVN) of the hypothalamus; their axons terminate in the medial portion of the external 21 . . . . 23 23 . . 24 . . . . . . . . 24 25 25 26 27 27 27 27 . . . . . . . . layer of the median eminence (Guillemin, 1978). Originally discovered in the hypothalamus, consistent with its classical role as a hypothalamic hypophysiotrophic factor, TRH is now known to be distributed extensively in extrahypothalamic brain structures (Winokur & Utiger, 1974; Yarbrough, 1979) and in other organs and tissues (Lechan, 1993). Similarly, receptors for TRH are found throughout the central and peripheral nervous system as well as in other organs and tissues (Sun et al., 2003). The widespread distribution of TRH and its receptors suggests other important functions for this tripeptide, including possible critical interactions with other biological systems (Gary et al., 2003; Yarbrough et al., 2007). The TRH receptors (TRH-R) belong to the seven transmembrane-spanning, G protein-coupled membrane receptor family (Sun et al., 2003). Two receptor isoforms, TRH receptor R1 (TRH-R1) and TRH receptor R2 (TRH-R2) have been identified (Gershengorn & Osman, 1996). In the brainstem, TRH-R1 has been shown to be present in the dorsal motor nucleus of the vagus (DMV) and the nucleus tractus solitarius (NTS), while TRH-R2 has been localized to the reticular formation, dorsal tegmental nucleus and spinal trigeminal nucleus (Heuer et al., 2000). TRH signaling occurs mainly via the phosphatidylinositol–calcium–protein kinase C transduction pathway, with subsequent elevations in intracellular calcium, and modulation of K+ channel conductance (Gershengorn & Osman, 1996). Notably, increasing evidence (Mellado et al., 1999; Montagne et al., 1999; Matre et al., 2003) supports the distribution of TRH and TRH receptors in the immune system and a number of studies provide data supporting potential interactions of TRH with the immune system, even at the level of regulation of transcription. 2. The expression of thyrotropin-releasing hormone and thyrotropin-releasing hormone-receptor in the immune system In addition to its classical function as a hypothalamic hypophysiotrophic factor, the widespread distribution of TRH and its receptors suggests that the tripeptide plays important roles in other systems. TRH has been identified throughout the CNS, including retina and spinal cord (Martino et al., 1980; Gary et al., 2003). TRH immunoreactivity has been detected in several peripheral tissues. Polymerase chain reaction (PCR) amplification analyses detected the expression of TRH in testes, adrenal glands, lymphoid organs, thymus, and spleen (Montagne et al., 1999). Immunohistochemistry analyses of rat adrenal gland extracts showed that TRH identified in this tissue is synthesized in mast cells (Montagne et al., 1997). It is possible that TRH identified in other peripheral tissues may, in fact, be synthesized there in the cells of the immune system. Similar to the expression of TRH, the expression of TRH receptors has been detected in several extrahypothalamic brain structures and peripheral tissues (Sun et al., 2003). TRH receptors have been detected in hematopoietic tissues related to the immune system, including thymus, bone marrow and lymphoid tissue (Sun et al., 2003). Northern blot analyses have identified TRH-R mRNA in immune cells (Raiden et al., 1995). Western blot analyses of extracts of rat lymphoid organs showed expression of TRH- 22 J. Kamath et al. / Pharmacology & Therapeutics 121 (2009) 20–28 R in thymus, mesenteric lymph nodes and spleen extracts (Mellado et al., 1999; Montagne et al., 1999). Expression analyses of rat tissues using monoclonal anti-TRH specific receptor antibodies have detected the presence of TRH receptors in several peripheral tissues related to the immune system, including thymus and lymphoid tissues (Fukusumi et al., 1995; Wang et al., 1997; Mellado et al., 1999; Bilek, 2000; Yamada et al., 2000). Analysis of human peripheral blood with these monoclonal antibodies detected TRH receptor expression in non-activated and phytohemagglutinin-activated T and B lymphocytes (Mallado et al., 1999). TRH receptor expression has been detected in both peripheral blood mononuclear cells (PBMC)-derived and in tonsil-derived B and T cells (Mallado et al., 1999). Altogether, it appears that TRH and its receptors exist and function as autocrine/paracrine systems in the immune system and other peripheral tissues and organs, perhaps analogous to its extrahypothalamic neurotransmitter/ neuromodulatory networks in the CNS. TRH may also influence the immune network indirectly. The tripeptide stimulates TSH (and thyroid hormones) and PRL, and both TSH and PRL have robust immunomodulatory properties (Kelley et al., 2007). Many different cells of the immune network have been shown to produce TSH. These include T cells, B cells, splenic dendritic cells, bone marrow hematopoietic cells, intestinal epithelial cells and lymphocytes (Klein, 2006). TRH was reported to stimulate the release of TSH from immune cells, and this effect was completely blocked by triiodothyronine (T3) administration (Komorowski et al., 1993). The presence of TSH receptors has been documented on multiple cells of the immune system, including lymphoid and myeloid cells, on select immune cell populations in the bone marrow, and on intestinal T cells (Klein, 2003). Similarly, expression of PRL has been shown in many different types of immune cells, and PRL can be produced by T lymphocytes and other cells of the immune system (Ben-Jonathan et al., 1996). Detailed discussions of interactions of TSH, thyroid hormones, and PRL with the immune system have been provided by Klein (2006) and Yu-Lee (2002). 3. In vitro evidence for interactions between thyrotropin-releasing hormone and the immune system Several studies have investigated the role of TRH within the immune system. Lesnikov et al. (1992) reported that a stereotactic lesion of the anterior hypothalamic area in mice produced rapid involution of the thymus and a reduction of lymphocytes in peripheral blood. This effect was prevented by the post-operational administration of TRH or melatonin and seemed to reflect direct activity of TRH on thymic targets or binding sites on lymphocytes. Consistent with this evidence, TRH has been reported to increase thymocyte cell proliferation in rats (Pawlikowski et al., 1992; Winczyk & Pawlikowski, 2000) and has been shown to antagonize the involution of the thymus produced by prednisolone (Pierpaoli & Yi, 1990). Thymectomy in pubertal rats resulted in significant depression of both TRH and TSH concentrations (Serebrov et al., 1992). TRH receptors have been detected on rat splenocytes (Raiden et al., 1995), and TRH has been shown to stimulate splenocyte proliferation (Raiden et al., 1995). Conversely, Kunert-Radek et al. (1991) reported suppression of spontaneous splenocyte proliferation by TRH. Studies in athymic mice indicate that TRH and TSH significantly influence the development of lymphoid cells associated with intestinal intraepithelial lymphocytes (IEL) (Wang & Klein, 1995). TRH receptors have been detected on IEL, and these receptors have been reported to be involved in the synthesis and secretion of TSH from intestinal T cells (Wang et al., 1997). Mice with congenitally mutant TSH receptors have been found to have a selectively impaired intestinal T cell repertoire (Wang et al., 1997). In vitro studies of effects of TRH on immune system function have suggested both stimulatory and inhibitory effects (Pawlikowski et al., 1994). For example, at low concentrations, TRH enhanced the T cellindependent antigen-induced antibody response via the production of TSH (Kruger et al., 1989). In contrast, Hart et al. (1990) demonstrated a decrease in IgG production and inhibition of monocytes by TRH. Moreover, Grasso et al. (1998) reported a stimulatory effect of TRH on in vitro interferon gamma (IFN-γ) production by human PBMCs. In contrast, human whole blood cells stimulated by mitogen, when incubated with TRH and imipramine, showed suppression of IFN-γ and interleukin-10 (IL-10) production (Kubera et al., 2000). Matre et al. (2003) established a novel functional link between TRH and the immune system by providing evidence that the human TRH-R1 receptor is transcriptionally regulated by the hematopoietic transcription factor c-Myb. In summary, TRH interacts with the immune system both during its development and in its fully developed state. These interactions are bidirectional and occur at all levels of the HPT axis. The effects of TRH on the immune system can be either stimulatory or inhibitory and are statedependent. It is important to note that these interactions have been investigated, to date, in the normal or inflammatory state; they have not been evaluated in the immunosuppressed state. The emergence of evidence suggesting interactions of these systems at the level of transcriptional regulation/signal transduction provides an opportunity to identify novel links as well as potential therapeutic targets. 4. In vivo evidence for interactions between thyrotropin-releasing hormone and the immune system Similar to the in vitro evidence, the in vivo evidence suggests both stimulatory and inhibitory interactions between TRH and the immune system. 4.1. Effects of thyrotropin-releasing hormone on the immune system Consistent with the in vitro evidence, intravenous TRH showed stimulatory effects on IFN-γ production in five normoprolactinemic women (Grasso et al., 1998). In healthy controls, intravenous TRH led to an increase in interleukin-2 (IL-2) concentrations (Komorowski et al., 1994; Trejbal et al., 2001), while in patients with hypothyroidism (with high baseline IL-2 concentrations), intravenous TRH caused a decrease in IL-2 concentrations (Trejbal et al., 2001). Studies conducted in animal models as well as in humans have suggested a role for TRH interactions in the pathophysiology of specific disorders involving changes in the immune system. Preliminary results have suggested a therapeutic potential for TRH analogs in the treatment of these disorders. TRH exerted a powerful protective effect in mice challenged with encephalomyelitis virus (Pierpaoli & Yi, 1990). It decreased the intensity of fungal invasion, decreased mortality rate, and increased survival time in a mouse model of experimental candidosis (Błaszkowska et al., 2004). Shimanko et al. (1992) reported protective effects of TRH in the treatment of edematous and destructive forms of acute pancreatitis. Intravenous and intra-lymphatic vessel administration of TRH in 15 patients with acute pancreatitis led to decreased edema and decreased amylasemia (Shimanko et al.,1992). In a series of experiments in rats, Yoneda et al. (2003, 2005a) reported stimulation of hepatic and pancreatic blood flow with microinjections of a TRH analog in the dorsal vagal complex. The stimulatory effect on hepatic blood flow was completely blocked by left cervical and hepatic branch vagotomy but not by right cervical vagotomy (Yoneda et al., 2003). Similarly stimulation of pancreatic blood flow was blocked by cervical vagotomy on the side of microinjection but not on the opposite side or by subdiaphragamtic vagotomy (Yoneda et al., 2005a). The effect was also blocked by pretreatment with atropine or N(G)-nitro-Larginine-methyl-ester (L-NAME), suggesting involvement of vagalcholinergic and nitric oxide-dependent pathways (Yoneda et al., 2005a). The same group reported protective effects of a centrally administered TRH analog on cerulin-induced acute pancreatitis in rats and blocking of this protective effect by subdiaphragmatic vagotomy or by pretreatment with L-NAME (Yoneda et al., 2005b). Taché et al. (2006) reported similar effects of central TRH administration on gastric J. Kamath et al. / Pharmacology & Therapeutics 121 (2009) 20–28 function. TRH injected into the DMN or cisterna magna increased vagal efferent discharge, activated cholinergic neurons in gastric submucosal and myenteric plexuses and induced a vagal-dependent, atropinesensitive stimulation of gastric secretory and motor functions. TRH antibody or TRH-R1 oligodeoxynucleotide antisense pretreatment in the DMN or cisterna magna completely abolished this effect (Taché et al., 2006). A TRH analogue was shown to induce gastric hyperemia via degranulation of mast cells (Kawakubo et al., 2005; Santos et al., 1996). Central TRH administration was shown to induce acute gastric lesions via vagal stimulation of ulcerogenic factors (acid, pepsin, motility, histamine) (Taché & Yoneda, 1993; Stephens et al., 1988). In the same set of experiments TRH was shown to have a cytoprotective effect against ethanol-induced gastric lesions by vagal-cholinergic stimulation of protective factors (prostaglandin, increased blood flow) (Taché & Yoneda, 1993). 4.2. Effects of cytokines on thyrotropin-releasing hormone systems The immune system, mainly involving cytokines, similarly seems to exert stimulatory or inhibitory effects on TRH systems. Prepro-TRH mRNA levels did not change in the acute phase of the lipopolysaccharide (LPS)-induced model of inflammation (Boelen et al., 2004). However, a significant increase in the expression of type 2 deiodinase (D2) mRNA was seen in the hypothalamus. The authors suggested that this enhanced D2 activity is a precursor for decreased hypothalamic TRH via increased local T3 generation due to negative feedback. This was confirmed in subsequent experiments conducted by Boelen et al. (2006) and by Pekary et al. (2007). Prepro-TRH mRNA concentrations significantly decreased 48 h after LPS administration in the PVN, and this correlated with increased expression of the pro-inflammatory cytokine interleukin-1 beta (IL-1β) in the hypothalamus (Boelen et al., 2006; Pekary et al., 2007). Boelen et al. (2006) noted that the D2 promoter region contains multiple nuclear factor (NF-kB) binding sites, suggesting a novel interaction at the level of transcriptional regulation between the immune system and TRH in the hypothalamus. Multiple pro-inflammatory cytokines have been tested for their effects on the HPT axis. Notably, these cytokines affect the HPT axis at multiple levels, leading to decreases in TRH expression, plasma TSH and thyroid hormone concentrations. However, the most dramatic effect seems to be on hypothalamic TRH expression. Continuous intraperitoneal infusion of interleukin-1 (IL-1) led to a 73% decrease in hypothalamic pro-TRH mRNA concentrations (van Haasteren et al., 1994). Similarly, a single intravenous injection of tumor necrosis factor-alpha (TNF-α; cachectin) reduced hypothalamic TRH (Pang et al.,1989), and increasing daily doses of TNF led to further significant reduction in hypothalamic TRH concentrations (Pang et al., 1989). IL-6 has been shown to inhibit TRH-stimulated PRL secretion and has also been shown to inhibit TRH-stimulated free cytosolic calcium increase (Schettini et al., 1991). In summary, the in vivo evidence, like the in vitro evidence, demonstrates bi-directional interactions between TRH and the immune system, occurring at all levels of the HPT axis. Evidence supports the notion that these interactions (stimulatory vs. inhibitory) may be statedependent, suggesting a homeostatic role for TRH in these interactions. However, just as with the in vitro evidence, effects of cytokines on TRH and on the HPT axis have been evaluated only in the normal or inflammatory state. A homeostatic role of TRH in these interactions can be conclusively established only after evaluations in the immunosuppressed state. 4.3. Role for thyrotropin-releasing hormone in the regulation of immune responses In contrast to the LPS model of nonspecific inflammation (T-cell independent response), where an immediate suppression of TRH is seen, the T-cell dependent antigens, i.e. sheep red blood cells (SRBC), elicited a rapid increase in hypothalamic TRH and pituitary TRH-R 23 mRNAs in the early phase (4–24 h post immunization). Notably, a decrease in levels similar to the LPS-induced, i.e. T-cell independent, response followed this initial rise (Perez et al., 1999). The initial increase in TRH was accompanied by a rise in plasma PRL levels. Intracerebroventricular injection of antisense oligonucleotide complementary to rat TRH mRNA resulted in a significant inhibition of specific antibody production and concomitant inability to produce the peak in plasma PRL levels in this model. This suggests that the T celldependent immune response and clonal expansion of T cells for appropriate antibody generation is critically dependent on the early activation of TRH (Perez et al., 1999). It is unclear which pathways mediate this early rise in hypothalamic TRH seen in the T-cell dependent response. It is possible that other neuromodulators (e.g., NPY, CART, glutamate, and vasopressin), which are activated during this immune response, may play roles in this early rise in TRH (Wittman, 2008). Evidence suggests that this early increase in TRH is then overridden by the direct suppressive effects of proinflammatory cytokines on TRH in the PVN, as in the LPS model. In summary, evidence also supports a critical role of TRH in the T-cell dependent immune response. Further exploration in this arena may lead to other therapeutic applications. 5. Connections between thyrotropin-releasing hormone and the immune system The delineation of two paradigms pertaining to interactions between the CNS, the immune system and the endocrine system has significantly advanced the field of psychoimmunology. One of these paradigms pertains to interactions between these systems in the LPS-induced sickness response. The second paradigm involves interactions between these systems to control gastric function and control of other visceral organs. We have already reviewed some evidence for a potential role for TRH in both of these paradigms. To avoid repetition, we will now review only the most critical in vivo evidence. We first describe the dynamic interactions between the CNS, the immune system and the endocrine system, and then elucidate a potential role for TRH in these interactions based on the evidence presented. Finally, we establish a foundation for TRH-based therapeutics in specific illnesses. Gary et al. (2003) were the first to provide an anatomical and functional framework to conceptualize diverse TRH pathways and TRH-mediated physiological and behavioral effects. The four distinct yet functionally integrated systems described by Gary et al. (2003) and Yarbrough et al. (2007) provided a framework for the authors to propose a pivotal role for TRH in the regulation of CNS homeostasis. In the current section, we delineate a role for TRH in a set of interactions that integrate the immune system with the CNS and the endocrine system. On the basis of the evidence provided, we propose that some of the physiological and behavioral events observed in disorders of immune function are mediated by effects exerted on TRH systems. 5.1. Anatomical framework for thyrotropin-releasing hormone–immune system interactions Current evidence suggests that the hypothalamus and brainstem serve as the epicenters of immune system interactions with other systems (Pavlov & Tracey, 2004). These two epicenters also serve to integrate overall brain responses to immune-derived signals from the periphery. This integration is achieved via multiple, mainly catecholaminergic, projections from these epicenters to the forebrain and other brain regions (Gaykema et al., 2007; Pavlov & Tracey, 2004). The critical areas within these epicenters that are involved in these interactions include: the PVN in hypothalamus and the NTS, the ventrolateral medulla (VLM) and the dorsal motor nucleus of vagus (DMN) in the brainstem. The PVN receives significant, mainly catecholaminergic, input from the lower brainstem centers (i.e., NTS, VLM and DMN) (Sawchenko et al., 2000). Select neuromodulators (e.g., corticotropinreleasing hormone [CRH], neuropeptide Y, histamine, and leptin) also 24 J. Kamath et al. / Pharmacology & Therapeutics 121 (2009) 20–28 Fig. 1. Overview of documented TRH and immune system interactions. play important roles in the pathophysiology of LPS-induced sickness syndrome. For example, significant increases in CRH in the PVN are observed after LPS administration. TRH and TRH receptors have a strong presence in both the hypothalamic and brainstem centers. Neurons in the dorsal vagal complex, including the DMN, express TRH receptors and are innervated by TRH fibers originating from TRH synthesizing neurons localized exclusively in the brainstem nuclei (Bayliss et al., 1994; Lynn et al., 1991). These brainstem nuclei, which contain TRH synthesizing neurons, namely the raphe pallidus, raphe obscurus, and parapyramidal regions, also harbor vagal and sympathetic preganglionic motor neurons involved in thermal, cardiovascular, gastrointestinal, and pancreatic regulation (Wittman, 2008; Taché et al., 2006). 5.2. Experimental evidence for vagus-dependent thyrotropin-releasing hormone–immune system interactions As described earlier, TRH administration in specific brainstem areas has been shown to impact functions of several visceral organs via vagal afferent-efferent pathways. Morrow et al. (1995) showed that a TRH analog (RX-77368) administered in the DVC induced gastric contractility. Microinjection of IL-1 beta (IL-1β) in the DVC (along with RX-77368) completely blocked this effect. Intracisternal injection of an IL-1 receptor antagonist abolished the inhibitory effect of IL-1β on the TRH analoginduced gastric contractility (Morrow et al., 1995). Hermann and Rogers (1995) showed a similar inhibitory effect of another pro-inflammatory cytokine, TNF-α, on TRH stimulated gastric motility. Compared to the IL1β inhibitory effect (30–120 min postinjection), the TNF-α effect was both immediate (within 30 s) and longer lasting. The TNF-α inhibitory effect was dose-dependent and required an intact vagal pathway (Hermann & Rogers, 1995). Hermann et al. (1999) demonstrated a similar inhibitory effect by intravenous administration of LPS. This inhibitory effect of LPS on TRH-induced gastric motility was reversed when endogenous TNF-α production was selectively suppressed (Hermann et al., 1999). Additionally, intravenous injections of bethanechol, a peripheral cholinergic ago- nist, were still able to elicit usual increases in gastric motility in the LPS model. These experiments confirmed that the inhibition of TRH-induced gastric motility seen in the LPS model was due to the central effects of endogenously produced proinflammatory cytokines, primarily TNF-α. Behavioral and physiological responses to LPS are achieved by communication of cytokine signals via vagal afferents to brain stem centers and further to the hypothalamus via catecholaminergic and other brainstem pathways (Fig. 1). The brain stem and hypothalamic centers coordinate the responses of various neuromodulators including TRH to the cytokine signals. These responses are then communicated to the periphery via vagal efferents and probably other, so far, unknown pathways. 5.3. Experimental evidence for vagus-independent thyrotropin-releasing hormone–immune system interactions Vagal afferents clearly play a major role in the communication of immune signals from the periphery to the CNS. However, vagotomy Table 1 Comparison of cytokine-induced sickness effects with clinical effects of TRH Domains Cytokine-induced sickness modela Neurovegetative Fatigue, psychomotor retardation, sleep alterations, anorexia Cognitive Decreased attention and concentration, memory difficulties Affective Depressed mood, anxiety, anhedonia Somatic Pain, gastrointestinal disturbances In-vivo TRH effectsb Arousal, vigilance, reversal of sedation, improved motor function Improved cognition including improved memory Antidepressant effects, improved emotional lability Pain modulation, gastric cytoprotective effects a From animal models and human use of cytokines (see Dantzer and Kelley, 2007 and Raison et al., 2006 for details). b Based on the in vivo data in animal models and human use (see Gary et al., 2003 and Yarbrough et al., 2007 for details). J. Kamath et al. / Pharmacology & Therapeutics 121 (2009) 20–28 25 Fig. 2. TRH and immune system interactions in health and disease; focus on LPS/cytokine-induced sickness response. was shown to block the behavioral (i.e. decreased social exploration) effects of IL-1β injected intraperitonially but not intravenously, suggesting that there are alternative pathways by which the cytokines can mediate effects on the CNS (Bluthé et al., 1996). Similarly, intraperitoneal IL-1β or LPS-induced physiological and behavioral responses were only slightly attenuated in subdiaphragmatically vagotomized rats (Wieczorek et al., 2005). Porter et al. (1998) showed that neither the vagal nor the nonvagal (splanchnic) afferent nerves from the upper gut are necessary for the anorexia produced by intraperitoneal IL-1β and LPS. Similar to the vagal afferents, the brainstem catecholaminergic pathways play a role in the activation of hypothalamic CRH neurons during the LPS-induced immune response (Ericsson et al., 1994). However, Fekete et al. (2005) showed that these brainstem pathways are not required in the LPS-induced suppression of TRH in the PVN. In a comprehensive review, Wittman (2008) suggests that the brainstem–catecholamine neurons–PVN pathway is necessary for the increase in CRH, but not for the TRH suppression in the LPS paradigm. It is important to note that, despite their large molecular size, cytokines can be transported into the brain, endogenously synthesized in the brain, or transmit signals to the brain via mechanisms independent of vagal afferents (Pavlov & Tracey, 2004). Wittman (2008) suggests that the suppression of TRH in the PVN occurs as the result of direct effects of proinflammatory cytokines, which may involve negative feedback due to increased local T3 production, as discussed earlier (Boelen et al., 2006). From an evolutionary perspective, it may be important to have a direct pathway for suppression of TRH (Fig. 1). The suppression of TRH may be necessary for survival during an adaptive sickness response (Kelley et al., 2003). 5.4. Role of thyrotropin-releasing hormone in the pathophysiology of the inflammatory process Both in vitro and in vivo evidence suggests that cytokine signaling to the brain may involve direct suppression of TRH by the proinflammatory cytokines in key brain centers such as the PVN and the DVC. The findings reviewed above suggest a major role for TRH in the core pathophysiology of the LPS-induced immune response. Suppres- sion of TRH may represent one of the critical events during an inflammatory process. It may be the event that drives the behavioral changes (e.g. social withdrawal, fatigue) and physiological changes that are typical of the cytokine-induced sickness response. The numerous clinical actions of TRH and TRH analogs (see Gary et al., 2003 and Yarbrough et al., 2007 for comprehensive reviews) such as arousal induction, enhancing cognitive function, improving motor function, and increasing gastric motility seem to be opposite to what is observed in the cytokine-induced sickness behavior paradigm (Table 1). The suppression of TRH seen in the LPS/proinflammatory cytokine model is consistent with the behavioral and physiological effects seen in the cytokine-induced sickness model (Fig. 2). In summary, the accumulated evidence (Fig. 1 and Table 2) strongly suggests significant suppressive effects of pro-inflammatory cytokines on TRH systems throughout the HPT axis during an inflammatory process. The suppressive effects on TRH are observed both in the hypothalamus (PVN) and in the brainstem (DVC) via or independent of vagal pathways. The clinical actions of TRH (Table 1) combined with the strong suppressive effects of proinflammatory cytokines on TRH systems during an inflammatory process suggest a potential role for TRH-based therapeutics in certain inflammatory disorders (Fig. 2). 6. Thyrotropin-releasing hormone-based therapeutics in inflammatory disorders The recognition and delineation of “proinflammatory cytokineinduced sickness behavior” has advanced the field of psychoimmunology in many ways. Dantzer and Kelley (2007) suggest that the common symptoms of sickness driven by proinflammatory cytokines – fatigue, anorexia, sleepiness, withdrawal from social activities, gastric stasis, fever, aching joints – are part of a “relative homeostasis” as a survival response to the trigger (e.g. infection). This acute sickness response is no longer adaptive if it is out of proportion to the insult or is unnecessarily prolonged (Elenkov et al., 2005). Increasing evidence suggests that such out of proportion or prolonged sickness behavior occurs in many disorders and inflicts serious physical and emotional consequences. One 26 J. Kamath et al. / Pharmacology & Therapeutics 121 (2009) 20–28 possible cause is the use of cytokines (interferons or interleukins) to treat certain types of cancers and hepatitis C (Raison et al., 2006). Other causes include the autoimmune disorders (e.g. rheumatoid arthritis, psoriasis, multiple sclerosis, ankylosing spondilytis, and inflammatory bowel diseases) (Elenkov et al., 2005). The prolonged sickness syndrome also has been observed after chemotherapy or radiation for cancer and after myocardial infarction (Kelley et al., 2003; Elenkov et al., 2005). The fatigue and depression observed in a subpopulation of patients with these disorders has been associated with a chronic inflammatory process (Miller et al., 2008). A number of recent studies associated the idiopathic fatigue reported by cancer patients with increase in specific pro-inflammatory cytokine levels (Bower, 2007). Even local radiation treatments in otherwise healthy cancer patients can cause significant fatigue. This radiation-induced fatigue has also been associated with increased plasma pro-inflammatory cytokine concentrations in patients with cancer (Jacobsen & Thors, 2003). Marquette et al. (2003) showed that the hypothalamus and certain other areas of Table 3 Future investigations of TRH-immune system interactions (not an exhaustive list) In vitro In vivo in animal models Table 2 Experimental evidence of bi-directional interactions between TRH and immune system Stimulating effects – TRH stimulated thymocyte cell proliferation and inhibited prednisolone-induced thymus involutiona – TRH stimulated splenocyte proliferationa – Thymectomy caused depressed thyroid function and decreased TRH, TSH levels⁎ – TRH enhanced T cell dependent, antigen induced antibody response – TRH stimulated IFN-α production by human peripheral blood mononuclear cells In vivo in – Central TRH analogue administration caused gastric animal hyperemia via degranulation of mast models cells – Central TRH analogue administration increased gastric, pancreatic and hepatic blood flow – Central TRH analogue administration enhanced gastric motor and secretory function ⇒ showed ulcerogenic potential – T cell dependent antigen response: In the early phase (b 24 h) showed increased hypothalamic TRH levels (required for adequate antibody production) In vitro In vivo in – Intravenous TRH showed humans stimulatory effect on IFN-γ and IL-2 levels in healthy subjects a In developmental stages. Inhibitory effects – TRH suppressed spontaneous splenocyte proliferationa – TRH caused decreased IgG production and inhibited monocytes – Mitogen-stimulated human whole blood cells when incubated with TRH and imipramine showed suppression of IFN-γ and IL-10 production – TRH showed protective effect in mice challenged with encephalomyelitis virus – Centrally administered TRH analogue showed protective effects in cerulin-induced pancreatitis – Central TRH analogue administration ⇒ showed cytoprotective effects in ethanolinduced gastric ulcers – Central administration (in DVC) of IL-1β and TNF-α blocked TRHinduced gastric motility – Intravenous LPS administration blocked TRH-induced gastric motility – Intravenous LPS administration caused suppression of hypothalamic TRH – Intraperitoneal IL-1 and intravenous TNF-α caused suppression of hypothalamic pro TRH mRNA levels – Intravenous IL-6 inhibited TRHstimulated prolactin secretion – T cell dependent antigen response: In the late phase (N24 h) showed suppression of hypothalamic TRH levels – Intravenous TRH caused decreased IL-2 levels in patients with hypothyroidism with baseline high IL-2 levels – Intravenous and intralymphatic vessel administration of TRH showed therapeutic effects in the treatment of acute pancreatitis In vivo in humans – Investigation of TRH role in the development of organs/tissues with autocrine/paracrine TRH networks – Investigation of TRH role and source in the peripheral organs/ tissues with autocrine/paracrine TRH networks – Investigation of differential expression of TRH-R1 and TRH-R2 receptors in the immune system and in other peripheral organs with TRH networks – Effects of TRH on immune cells and on cytokine production in immunosuppressed state – Investigation of development of TRH system/HPT axis in the inflammatory vs. immunosuppressed state – Investigation of TRH system in the immunosuppressed state of the fully developed immune system – Behavioral and other effects of TRH in the inflammatory vs. immunosuppressed state – Behavioral and other effects of receptor specific TRH analogs (TRH-R1 vs. TRH-R2) on the immune system in general and in the inflammatory vs. immunosuppressed state – Investigation of TRH interactions with other critical neuromodulators (i.e. CRF, vasopressin, prolactin) in differential immune states – Investigation of TRH-induced immunomodulatory effect in select disorders (for example acute pancreatitis, spinal chord injury, and Alzheimer's disease) and its contribution to the therapeutic effects associated with TRH and its analogs in these disorders – Investigation of therapeutic effects of TRH and receptor specific TRH analogs in select animal models of inflammatory disorders – Investigation of immune system in the hyper vs. hypothyroid states – Investigation of TRH system/HPT axis in the immunosuppressed state – Investigation of therapeutic effects of TRH and its analogs in select inflammatory disorders the brain of partial-body irradiated rats show high levels of proinflammatory cytokines, specifically IL-1β, TNF-α, and IL-6. This increase in cytokine levels was prevented by vagotomy before irradiation, confirming the importance of the vagal pathway for cytokine signaling (Marquette et al., 2003). 6.1. Cytokine-induced sickness syndrome as a therapeutic target in inflammatory disorders Corticosteroids, known to suppress the inflammatory process, are frequently used in palliative treatment for their anti-fatigue, anticachexia and anti-anorexia effects (Shih & Jackson, 2007). The first identification of the “sickness syndrome” as a therapeutic target came from the behavioral data generated by the clinical use of TNF inhibitors (recombinant soluble form of TNF receptor). The introduction of these new anti-TNF drugs (etanercept, infliximab, and adalimumab) has significantly advanced the field of rheumatology. These drugs treat not only the specific aspects of an illness but also cause an overall improvement in patient functioning and quality of life. In clinical studies with these agents, patients reported less fatigue, improved physical function and better emotional and mental function (Nash & Florin, 2005). The “anti-sickness” effects of these agents have been reported in an array of autoimmune diseases including rheumatoid arthritis, ankylosing spondylitis, Wegeners granulomatosis, psoriasis, and inflammatory bowel diseases (Nash & Florin, 2005). In a recent pilot study, Monk et al. (2006) confirmed antifatigue effects of etanercept in cancer patients undergoing chemotherapy. Unfortunately, these agents have been associated with significant side effects. For some of these agents, the Food and Drug Administration has added a “black box” warning of risk of serious infections (Rychly & DiPiro, 2005). Thus, more specific therapeutic agents are needed to counteract the behavioral consequences of autoimmune disorders. TRH-based therapeutics may provide one such approach based on the hypothesis that some of the behavioral J. Kamath et al. / Pharmacology & Therapeutics 121 (2009) 20–28 symptoms of inflammatory disorders are due to the suppressive effects of cytokines on TRH systems. This hypothesis needs further evaluation in in vitro and in vivo settings. 6.2. Challenges and advances in the development of thyrotropin-releasing hormone-based therapeutics The development of TRH-based therapeutics has been hampered by the short half-life of TRH and its limited access to the CNS after peripheral administration. However, it is important to emphasize that, in contrast to the recent novel treatments for inflammatory disorders (i.e. TNF blockers) with potentially serious side effects, TRH has been in clinical use since 1974. It has a favorable safety record both in clinical use and in research studies, including studies in patients with serious illnesses (Gary et al., 2003; Yarbrough et al., 2007). Given the limitations of native TRH, it is important to utilize metabolically stable TRH analogs with better access to the CNS. Two TRH analogs, TA-0910 (Ceredist) and CG-3703, possessing these properties, have shown clinical promise (Gary et al., 2003). Ceredist has been used in Japan since 2001 for the treatment of spinocerebellar degeneration. Clinical investigations can be conducted to test these and related compounds for the treatment of behavioral and other aspects of certain inflammatory disorders (i.e. autoimmune diseases, inflammatory bowel diseases, cancer-related fatigue or depression, and Alzheimer's disease). Additionally, the development of new TRH analogs, especially receptor subtype selective compounds, represents an area of significant research potential. 7. Future investigations Future investigations should include evaluation of TRH-based therapeutics in inflammatory disorders, especially for the behavioral aspects of the sickness syndrome associated with these disorders. On a conceptual level, it is important to investigate whether the homeostatic role for TRH in the CNS put forth by Gary et al. (2003) extends to the regulation of the immune system. Studies to evaluate the differential roles of the two types of TRH receptors in TRH-immune system interactions are also a matter of high priority. As mentioned earlier, TRH and TRH analogs have shown clinical promise in certain disorders (Gary et al., 2003). Many of these disorders are associated with inflammatory processes, and immunomodulation may play a role in the therapeutic effects associated with TRH or its analogs. It is important to test this hypothesis in animal models of these disorders. Potential experiments to investigate these hypotheses are described in Table 3. 8. Conclusions TRH has numerous interactions with the immune system in the CNS as well as in the periphery and during multiple stages of development. These interactions can be direct or indirect, i.e. via other neuromodulators. A large body of in vitro and in vivo evidence supports a homeostatic role for TRH in its interactions with the immune system, extending the hypothesis previously proposed by Gary et al. (2003) and Yarbrough et al. (2007) of a homeostatic regulatory role for TRH to include effects on immune system function. The TRH-immune system homeostatic hypothesis states that TRHmediated mechanisms respond to many elements of the immune system and affect them in ways that tend to maintain or restore homeostasis. Some aspects of this hypothesis may be tested by the use of TRH or its congeners to treat patients with certain inflammatory diseases or to affect animal models of those diseases. The critical role of TRH in the cytokine-induced sickness paradigm presents excellent opportunities for further exploration of this hypothesis and provides promising targets for TRH-based therapeutics of immune systemrelated medical and psychiatric disorders. 27 References Ader, R. (1980). Presidential address: psychosomatic and psychoimmunologic research. Psychosom Med 42, 307−322. Bayliss, D. A., Viana, F., Kanter, R. K., Szymeczek-Seay, C. L., Berger, A. J., & Millhorn, D. E. (1994). Early postnatal development of thyrotropin-releasing hormone (TRH) expression, TRH receptor binding, and TRH responses in neurons of rat brainstem. J Neurosci 14(2), 821−833. Ben-Jonathan, N., Mershon, J., Allen, D., & Steinmetz, R. (1996). Extrapituitary prolactin: distribution, regulation, functions and clinical aspects. Endocr Rev 17, 639−669. Bilek, R. (2000). TRH-like peptides in prostate gland and other tissues. Physiol Res 49 (Suppl 1), 519−526. Błaszkowska, J., Pawlikowski, M., Komorowski, J., & Kurnatowski, P. (2004). Effect of thyroliberin on the course of experimental candidosis in mice. Mycoses 47(3–4), 115−120. Bluthé, R. M., Michaud, B., Kelley, K. W., & Dantzer, R. (1996). Vagotomy blocks behavioral effects of interleukin-1 injected via the intraperitoneal route but not via other systemic routes. Neuroreport 7(15–17), 2823−2827. Boelen, A., Kwakkel, J., Thijssen-Timmer, D. C., Alkemade, A., Fliers, E., & Wiersinga, W. M. (2004). Simultaneous changes in central and peripheral components of the hypothalamus–pituitary–thyroid axis in lipopolysaccharide-induced acute illness in mice. J Endocrinol 182(2), 315−323. Boelen, A., Kwakkel, J., Wiersinga, W. M., & Fliers, E. (2006). Chronic local inflammation in mice results in decreased TRH and type 3 deiodinase mRNA expression in the hypothalamic paraventricular nucleus independently of diminished food intake. J Endocrinol 191(3), 707−714. Bower, J. E. (2007). Cancer-related fatigue: links with inflammation in cancer patients and survivors. Brain Behav Immun 21(7), 863−871. Dantzer, R., & Kelley, K. W. (2007). Twenty years of research on cytokine-induced sickness behavior. Brain Behav Immun 21(2), 153−160. Elenkov, I. J., Iezzoni, D. G., Daly, A., Harris, A. G., & Chrousos, G. P. (2005). Cytokine dysregulation, inflammation and well-being. Neuroimmunomodulation 12(5), 255−269. Ericsson, A., Kovács, K. J., & Sawchenko, P. E. (1994). A functional anatomical analysis of central pathways subserving the effects of interleukin-1 on stress-related neuroendocrine neurons. J Neurosci 14(2), 897−913. Eskandari, F., & Sternberg, E. M. (2002). Neural-immune interactions in health and disease. Ann N Y Acad Sci 966, 20−27. Fekete, C., Singru, P. S., Sarkar, S., Rand, W. M., & Lechan, R. M. (2005). Ascending brainstem pathways are not involved in lipopolysaccharide-induced suppression of thyrotropin-releasing hormone gene expression in the hypothalamic paraventricular nucleus. Endocrinology 146(3), 1357−1363. Fukusumi, S., Ogi, K., Onda, H., & Hinuma, S. (1995). Distribution of thyrotropinreleasing hormone receptor mRNA in rat peripheral tissues. Regul Pept 57(2), 115−121. Gary, K. A., Sevarino, K. A., Yarbrough, G. G., Prange, A. J., Jr., & Winokur, A. (2003). The thyrotropin-releasing hormone (TRH) hypothesis of homeostatic regulation: implications for TRH-based therapeutics. J Pharmacol Exp Ther 305(2), 410−416. Gaykema, R. P., Chen, C. C., & Goehler, L. E. (2007). Organization of immune-responsive medullary projections to the bed nucleus of the stria terminalis, central amygdala, and paraventricular nucleus of the hypothalamus: evidence for parallel viscerosensory pathways in the rat brain. Brain Res 1130(1), 130−145. Gershengorn, M. C., & Osman, R. (1996). Molecular and cellular biology of thyrotropinreleasing hormone receptors. Physiol Rev 76(1), 175−191. Grasso, G., Massai, L., De Leo, V., & Muscettola, M. (1998). The effect of LHRH and TRH on human interferon-gamma production in vivo and in vitro. Life Sci 62(22), 2005−2014. Guillemin, R. (1978). Peptides in the brain: the new endocrinology of the neuron. Science 202, 390−402. Hart, R., Wagner, F., Steffens, W., Lersch, C., Dancygier, H., Duntas, L., et al. (1990). Effect of thyrotropin-releasing hormone on immune functions of peripheral blood mononuclear cells. Regul Pept 27(3), 335−342. Hermann, G., & Rogers, R. C. (1995). Tumor necrosis factor-alpha in the dorsal vagal complex suppresses gastric motility. Neuroimmunomodulation 2(2), 74−81. Hermann, G. E., Tovar, C. A., & Rogers, R. C. (1999). Induction of endogenous tumor necrosis factor-alpha: suppression of centrally stimulated gastric motility. Am J Physiol 276(1 Pt 2), R59−68. Heuer, H., Schäfer, M. K., O'Donnell, D., Walker, P., & Bauer, K. (2000). Expression of thyrotropin-releasing hormone receptor 2 (TRH-R2) in the central nervous system of rats. J Comp Neurol 428(2), 319. Jacobsen, P. B., & Thors, C. L. (2003). Fatigue in the radiation therapy patient: current management and investigations. Semin Radiat Oncol 13(3), 372−380. Kawakubo, K., Akiba, Y., Adelson, D., Guth, P. H., Engel, E., Taché, Y., et al. (2005). Role of gastric mast cells in the regulation of central TRH analog-induced hyperemia in rats. Peptides 26(9), 1580−1589. Keller, S. E., Schleifer, S. J., Liotta, A. S., Bond, R. N., Farhoody, N., & Stein, M. (1988). Stressinduced alterations of immunity in hypophysectomized rats. Proc Natl Acad Sci U S A 85(23), 9297−9301. Kelley, K. W., Bluthé, R. M., Dantzer, R., Zhou, J. H., Shen, W. H., Johnson, R. W., et al. (2003). Cytokine-induced sickness behavior. Brain Behav Immun 17(Suppl 1), S112−118. Kelley, K. W., Weigent, D. A., & Kooijman, R. (2007). Protein hormones and immunity. Brain Behav Immun 21(4), 384−392. Klein, J. R. (2003). Physiological relevance of thyroid stimulating hormone and thyroid stimulating hormone receptor in tissues other than the thyroid. Autoimmunity 36(6–7), 417−421. 28 J. Kamath et al. / Pharmacology & Therapeutics 121 (2009) 20–28 Klein, J. R. (2006). The immune system as a regulator of thyroid hormone activity. Exp Biol Med (Maywood) 231(3), 229−236. Komorowski, J., Stepień, H., & Pawlikowski, M. (1993). The evidence of thyroliberin/ triiodothyronine control of TSH secretory response from human peripheral blood monocytes cultured in vitro. Neuropeptides 25(1), 31−34. Komorowski, J., Stepien, H., & Pawlikowski, M. (1994). Increased interleukin-2 levels during standard TRH test in man. Neuropeptides 27, 151−156. Kruger, T. E. (1996). Immunomodulation of peripheral lymphocytes by hormones of the hypothalamus–pituitary–thyroid axis. Adv Neuroimmunol 6(4), 387−395. Kubera, M., Kenis, G., Bosmans, E., Jaworska-Feil, L., Lasoń, W., Scharpe, S., et al. (2000). Suppressive effect of TRH and imipramine on human interferon-gamma and interleukin-10 production in vitro. Pol J Pharmacol 52(6), 481−486. Kruger, T. E., Smith, L. R., Harbour, D. V., & Blalock, J. E. (1989). Thyrotropin:an endogenous regulator of the in vitro immune response. J Immunol 142(3), 744−747. Kunert-Radek, J., Pawlikowski, M., Stepien, H., & Janecka, A. (1991). Inhibitory effect of thyrotropin releasing hormone on spontaneous proliferation of mouse spleen lymphocytes in vitro. Biochem Biophys Res Commun 181(2), 562−565. Lechan, R. M. (1993). Update on thyrotropin-releasing hormone. Thyroid Today 16, 1−11. Lesnikov, V. A., Korneva, E. A., Dall'ara, A., & Pierpaoli, W. (1992). The involvement of pineal gland and melatonin in immunity and aging: II. Thyrotropin-releasing hormone and melatonin forestall involution and promote reconstitution of the thymus in anterior hypothalamic area (AHA)-lesioned mice. Int J Neurosci 62(1–2), 141−153. Lynn, R. B., Feng, H. S., Han, J., & Brooks, F. P. (1991). Gastric effects of thyrotropinreleasing hormone microinjected into the dorsal vagal nucleus in cats. Life Sci 48(13), 1247−1254. Marquette, C., Linard, C., Galonnier, M., Van Uye, A., Mathieu, J., Gourmelon, P., et al. (2003). IL-1beta, TNFalpha and IL-6 induction in the rat brain after partial-body irradiation: role of vagal afferents. Int J Radiat Biol 79(10), 777−785. Martino, E., Nardi, M., Vaudagna, G., Simonetti, S., Cilotti, A., Piunchera, A., et al. (1980). Thyrotropin-releasing hormone-like material in human retina. J Endocrinol Invest 3, 267−271. Matre, V., Høvring, P. I., Fjeldheim, A. K., Helgeland, L., Orvain, C., Andersson, K. B., et al. (2003). The human neuroendocrine thyrotropin-releasing hormone receptor promoter is activated by the haematopoietic transcription factor c-Myb. Biochem J 372(Pt 3), 851−859. Mellado, M., Fernández-Agulló, T., Rodríguez-Frade, J. M., San Frutos, M. G., de la Peña, P., Martínez-A, C., et al. (1999). Expression analysis of the thyrotropin-releasing hormone receptor (TRHR) in the immune system using agonist anti-TRHR monoclonal antibodies. FEBS Lett 28(451(3)), 308−314. Miller, A. H., Ancoli-Israel, S., Bower, J. E., Capuron, L., & Irwin, M. R. (2008). Neuroendocrine-immune mechanisms of behavioral comorbidities in patients with cancer. J Clin Oncol 26(6), 971−982. Monk, J. P., Phillips, G., Waite, R., Kuhn, J., Schaff, L. J., Otterson, G. A., et al. (2006). Assessment of tumor necrosis factor alpha blockade as an intervention to improve tolerability of dose-intensive chemotherapy in cancer patients. J Clin Oncol 24(12), 1852−1859. Montagne, J. J., Ladram, A., Grouselle, D., Nicolas, P., & Bulant, M. (1997). Thyrotropinreleasing hormone immunoreactivity in rat adrenal tissue is localized in mast cells. J Histochem Cytochem 45(12), 1623−1627. Montagne, J. J., Ladram, A., Nicolas, P., & Bulant, M. (1999). Cloning of thyrotropinreleasing hormone precursor and receptor in rat thymus, adrenal gland, and testis. Endocrinology 140(3), 1054−1059. Morrow, N. S., Quinonez, G., Weiner, H., Taché, Y., & Garrick, T. (1995). Interleukin-1 beta in the dorsal vagal complex inhibits TRH analogue-induced stimulation of gastric contractility. Am J Physiol 269(2 Pt 1), G196−202. Nagy, E., & Berczi, I. (1978). Immunodeficiency in hypophysectomized rats. Acta Endocrinol 89(3), 530. Nash, P. T., & Florin, T. H. (2005). Tumour necrosis factor inhibitors. Med J 183(4), 205−208. Nillni, E. A., & Sevarino, K. A. (1999). The biology of pro-thyrotropin-releasing hormonederived peptides. Endocr Rev 20(5), 599−648. Pang, X. P., Hershman, J. M., Mirell, C. J., & Pekary, A. E. (1989). Impairment of hypothalamic-pituitary–thyroid function in rats treated with human recombinant tumor necrosis factor-alpha (cachectin). Endocrinology 125(1), 76−84. Pavlov, V. A., & Tracey, K. J. (2004). Neural regulators of innate immune responses and inflammation. Cell Mol Life Sci 61(18), 2322−2331. Pawlikowski, M., Stepien, H., & Komorowski, J. (1994). Hypothalamic-pituitary–thyroid axis and the immune system. Neuroimmunomodulation 1(3), 149−152. Pawlikowski, M., Zerek-Mełeń, G., & Winczyk, K. (1992). Thyroliberin (TRH) increases thymus cell proliferation in rats. Neuropeptides 23(3), 199−202. Pekary, A. E., Stevens, S. A., & Sattin, A. (2007). Lipopolysaccharide modulation of thyrotropin-releasing hormone (TRH) and TRH-like peptide levels in rat brain and endocrine organs. J Mol Neurosci 31(3), 245−259. Perez, C. C., Penaleva, R., Paez, P. M., Renner, U., Reul, J. M., Stalla, G. K., et al. (1999). Early activation of thyrotropin-releasing-hormone and prolactin plays a critical role during a T cell-dependent immune response. Endocrinology 140(2), 690−697. Pierpaoli, W., & Yi, C. (1990). The involvement of pineal gland and melatonin in immunity and aging. I. Thymus-mediated, immunoreconstituting and antiviral activity of thyrotropin-releasing hormone. J Neuroimmunol 27(2–3), 99−109. Porter, M. H., Hrupka, B. J., Langhans, W., & Schwartz, G. J. (1998). Vagal and splanchnic afferents are not necessary for the anorexia produced by peripheral IL-1beta, LPS, and MDP. Am J Physiol 275(2 Pt 2), R384−389. Raiden, S., Polack, E., Nahmod, V., Labeur, M., Holsboer, F., & Arzt, E. (1995). TRH receptor on immune cells: in vitro and in vivo stimulation of human lymphocyte and rat splenocyte DNA synthesis by TRH. J Clin Immunol 15(5), 242−249. Raison, C. L., Capuron, L., & Miller, A. H. (2006). Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol 27(1), 24−31. Rychly, D. J., & DiPiro, J. T. (2005). Infections associated with tumor necrosis factor-alpha antagonists. Pharmacotherapy 25(9), 1181−1192. Santos, J., Saperas, E., Mourelle, M., Antolín, M., & Malagelada, J. R. (1996). Regulation of intestinal mast cells and luminal protein release by cerebral thyrotropin-releasing hormone in rats. Gastroenterology 111(6), 1465−1473. Sawchenko, P. E., Li, H. Y., & Ericsson, A. (2000). Circuits and mechanisms governing hypothalamic responses to stress: a tale of two paradigms. Prog Brain Res 122, 61−78. Schettini, G., Grimaldi, M., Landolfi, E., Meucci, O., Ventra, C., Florio, T., et al. (1991). Role of interleukin-6 in the neuroendocrine system. Acta Neurol (Napoli) 13(4), 361−367. Serebrov, V., Zobnina, M. N., & Tikhonova, N. M. (1992). Structural–functional state of the thyroid gland after thymectomy. Biull Eksp Biol Med 114(9), 329−332. Shih, A., & Jackson, K. C., 2nd (2007). Role of corticosteroids in palliative care. J Pain Palliat Care Pharmacother 21(4), 69−76. Shimanko, I. I., Limarev, V. M., Ashmarin, I. P., Lelekova, T. V., & Sanzhieva, L. T. (1992). The use of thyrotropin-releasing hormone in clinical practice as a lymphatic stimulator in the treatment of acute pancreatitis. Khirurgiia (Mosk)(1), 64−66. Stephens, R. L., Ishikawa, T., Weiner, H., Novin, D., & Tache, Y. (1988). TRH analogue, RX 77368, injected into dorsal vagal complex stimulates gastric secretion in rats. Am J Physiol 254(5 Pt 1), G639−643. Sternberg, E. M. (1995). Neuroendocrine factors in susceptibility to inflammatory disease: focus on the hypothalamic-pituitary–adrenal axis. Horm Res 43(4), 159−161. Sun, Y., Lu, X., & Gershengorn, M. C. (2003). Thyrotropin-releasing hormone receptors — similarities and differences. J Mol Endocrinol 30(2), 87−97. Taché, Y., & Yoneda, M. (1993). Central action of TRH to induce vagally mediated gastric cytoprotection and ulcer formation in rats. J Clin Gastroenterol 17(Suppl 1), S58−63. Taché, Y., Yang, H., Miampamba, M., Martinez, V., & Yuan, P. Q. (2006). Role of brainstem TRH/TRH-R1 receptors in the vagal gastric cholinergic response to various stimuli including sham-feeding. Auton Neurosci 125(1–2), 42−52. Trejbal, D., Petrasova, D., & Wagnerova, H. (2001). Prolactin and interleukin 2 concentrations before and after i.v. TRH application in primary hypothyroidism and in controls. Bratisl Lek Listy 102(9), 417−419. van Haasteren, G. A., van der Meer, M. J., Hermus, A. R., Linkels, E., Klootwijk, W., Kaptein, E., et al. (1994). Different effects of continuous infusion of interleukin-1 and interleukin-6 on the hypothalamic-hypophysial-thyroid axis. Endocrinology 135(4), 1336−1345. Wang, J., & Klein, J. R. (1995). Hormonal regulation of extrathymic gut T cell development: involvement of thyroid stimulating hormone. Cell Immunol 161(2), 299−302. Wang, J., Whetsell, M., & Klein, J. R. (1997). Local hormone networks and intestinal T cell homeostasis. Science 275(5308), 1937−1939. Winokur, A., & Utiger, R. D. (1974). Thyrotropin-releasing hormone: regional distribution in rat brain. Science 185, 265−266. Wittman, G. J. (2008). Regulation of hypophysiotropic corticotropin-releasing hormone and thyrotropin-releasing hormone-synthesizing neurons by brainstem catecholaminergic neurons. Neuroendocrinol 0(7), 952−960. Wieczorek, M., Swiergiel, A. H., Pournajafi-Nazarloo, H., & Dunn, A. J. (2005). Physiological and behavioral responses to interleukin-1beta and LPS in vagotomized mice. Physiol Behav 85(4), 500−511. Winczyk, K., & Pawlikowski, M. (2000). Time of day-dependent effects of thyroliberin and thyrotropin on thymocyte proliferation in rats. Neuroimmunomodulation 7(2), 89−91. Yamada, M., Shibusawa, N., Hashida, T., Ozawa, A., Monden, T., Satoh, T., et al. (2000). Expression of thyrotropin-releasing hormone (TRH) receptor subtype 1 in mouse pancreatic islets and HIT-T15, an insulin-secreting clonal beta cell line. Life Sci 66(12), 1119−1125. Yarbrough, G. G. (1979). On the neuropharmacology of thyrotropin releasing hormone (TRH). Prog Neurobiol 12(3–4), 291−312. Yarbrough, G. G., Kamath, J., Winokur, A., & Prange, A. J., Jr (2007). Thyrotropin-releasing hormone (TRH) in the neuroaxis: therapeutic effects reflect physiological functions and molecular actions. Med Hypotheses 69(6), 1249−1256. Yoneda, M., Hoshimoto, T., Nakamura, K., Tamori, K., Yokohama, S., Kono, T., et al. (2003). Thyrotropin-releasing hormone in the dorsal vagal complex stimulates hepatic blood flow in rats. Hepatology 38(6), 1500−1507. Yoneda, M., Goto, M., Nakamura, K., Yokohama, S., Kono, T., Tanamo, M., et al. (2005). Thyrotropin-releasing hormone in the dorsal vagal complex stimulates pancreatic blood flow in rats. Regul Pept 131(1–3), 74−81. Yoneda, M., Goto, M., Nakamura, K., Shimada, T., Hiraishi, H., Terano, A., et al. (2005). Protective effect of central thyrotropin-releasing hormone analog on ceruleininduced acute pancreatitis in rats. Regul Pept 125(1–3), 119−124. Yu-Lee, L. Y. (2002). Prolactin modulation of immune and inflammatory responses. Recent Prog Horm Res 57, 435−455.