Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Magnetic circular dichroism wikipedia , lookup
Marcus theory wikipedia , lookup
Auger electron spectroscopy wikipedia , lookup
Molecular Hamiltonian wikipedia , lookup
Metastable inner-shell molecular state wikipedia , lookup
Gamma spectroscopy wikipedia , lookup
Physical organic chemistry wikipedia , lookup
Rotational spectroscopy wikipedia , lookup
George S. Hammond wikipedia , lookup
Eigenstate thermalization hypothesis wikipedia , lookup
Mössbauer spectroscopy wikipedia , lookup
Heat transfer physics wikipedia , lookup
X-ray photoelectron spectroscopy wikipedia , lookup
Ultraviolet–visible spectroscopy wikipedia , lookup
Franck–Condon principle wikipedia , lookup
Rotational–vibrational spectroscopy wikipedia , lookup
X-ray fluorescence wikipedia , lookup
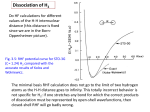
Dissociation energy of the C-H bond in chloroform Purpose This experiment is to determine the dissociation energy of the carbonhydrogen bond in chloroform. Dissociation energy will be calculated from fundamental and overtone vibrational energies measured with infrared and near-infrared spectrophotometers. The dissociation energy will also be calculated with quantum-chemical methods. Introduction Chloroform and other small chlorinated molecules are important organic solvents. They are also important in atmospheric chemistry. Hydrogen abstraction is an important initial step in degradation of chlorinated molecules in the atmosphere, and the rate of hydrogen abstraction is strongly correlated with the C-H bond strength. Homolytic bond cleavage is a simple process to write: Cl3C-H → Cl3C· + ·H (1) The energy of that reaction is the bond dissociation energy. The products of the reaction are highly reactive radicals, making experimental measurement difficult. Experimental methods and results were reviewed by McMillen and Golden.1 Quantum calculations are also difficult because the number of bonds change in the reaction (electron correlation is important) and spin changes: a singlet reactant yields two doublet products. Although bond dissociation energy is an important concept, it is not easy to measure experimentally. McGivern, et al.,.2 say that bomb calorimetry "is the primary method for the experimental determination of bond dissociation energies." Quantum-chemical calculations offer an attractive alternative to experiments, but are subject to uncertainty. Gribov, et al.,3 warn readers that "When comparing bond dissociation energies determined by different experimental and theoretical methods, it should be kept in mind that none of these methods is absolute." Total agreement between spectroscopic and quantum-chemical dissociation energy is not to be expected. The experimental method used in this lab is to measure vibrational energies of the C-H stretching vibration in liquid chloroform. The fundamental frequency will be measured with a Fourier-transform infrared instrument. Overtones will be measured with a scanning near-infrared spectrophotometer. Differences of vibration frequencies will be extrapolated to the dissociation limit, following the Birge-Sponer method. The experimental procedure follows that published by Myrick, et al.4 Quantum-chemical calculations will be done with density functional theory (DFT), an electronic-structure theory that includes correlation energy. The "hybrid" functional B3LYP will be used. It is hybrid in that it mixes some Hartree-Fock exchange energy with pure density functional theory. It is the most-used functional for quantum chemistry of small molecules. CHbond.odt 1 Theory Vibrational spectroscopy is related to dissociation energy through anharmonicity. Consider the C-H stretching vibration. Transitions observed in infrared and near-infrared spectroscopy give energy differences between the v=0 energy level and a higher-v level, where v is the vibrational ν, quantum number. For a peak observed at wavenumber , the transition energy is Δ E=hc ~ where h is Planck's constant and c is the speed of light. The energy calculated in that way is energy per photon, and may be converted to energy per mole by multiplying by Avogadro's number. For a peak observed at wavelength , the transition energy is Δ E =hc / λ , likewise per photon. The diagram at right shows an anharmonic potential energy surface. A surface of this sort describes the C-H bond in chloroform. Vibrational energy levels are drawn and vibrational quantum numbers v=0, 1, 2, 3, 4 and 5 are shown. Vibrational transitions are shown. The 0-1 transition is the fundamental transition and will be observed in the infrared spectrum. The 0-2, 0-3, 0-4, and 0-5 transitions are the first, second, third and fourth overtones. The dissociation energy, D0, is the convergence limit of the overtones. The progression of overtone energies can be analyzed in terms of anharmonicity as follows. For Figure 1: anharmonic potential energy surface this derivation, suppose all energies are in wavenumber units, cm-1. The energy of the vibrational level having quantum number v is G(v). The expression for G(v) is given below including only the first anharmonic term, which is sufficient for the experiment. 1 ~ 1 − νe x e v + 2 2 ( ) G (v ) = ~ νe v + 2 ( ) (2) The energy difference between two adjacent levels, v and v+1, is denoted Δ G v +1/2 . The figure at right (taken from Peter Bernath's book6) shows the expected progression of Δ Gv +1/ 2 . Δ G v+ 1/ 2 ≡ G (v+ 1) − G (v ) ΔG =~ ν − 2~ ν x (v +1) v+ 1/ 2 e e e (3) Figure 2: vibrational progression Equation 3 has the form of a straight line. Δ G v+1/ 2 = b + m(v +1) CHbond.odt (4) 2 ~ When Δ G v +1/2 is graphed versus (v+1), the y-intercept should be b= ν e and the slope m = −2 ~ ν e xe . The x-intercept, minus 1, is the quantum number at which dissociation occurs, vd. The dissociation energy D0 is then easily calculated. D 0 = G (v d ) − G (0) = v d ~ ν e − v d (v d +1)~ νe x e (5) In terms of the slope and intercept, Myrick, et al., give the following convenient formula:4 D0 = − b( b+m) 2m (6) Calculated in this way, D0 will have units of wavenumbers. It can be converted to kJ/mol by multiplying by hc and by Avogadro's number, and converting Joules to kiloJoules. Two results from the spectroscopic experiment can readily be compared to results of quantum calculations. Those results are the dissociation energy D0 , and the harmonic vibration ~ frequency ν e . Quantum-chemical calculations of ~ ν e and D0 are straightforward. The plan is to calculate the energies of the products and reactants of the dissociation reaction (equation 1), and then take the difference, products minus reactants. Both HCCl3 and ·CCl3 should be geometry-optimized. Their energies will also be corrected for zero-point vibration. ν ν 6 ~ 9 ~ D 0 = E ( H ) + E (CCl 3 )+ ∑ i =1 CCl 3,i 2 ( − E (HCCl 3 )+ ∑ i=1 HCCl 3,i 2 ) (7) When using the concept of equation 7, unit conversions will be necessary. Note that 1 Hartree = 219470 cm-1 = 2625.5 kJ/mol Because the products and reactants have different numbers of electron pairs, a correlated computational method is essential. We will use density functional theory, specifically the B3LYP functional. Correlated methods work best with large basis sets. We will use the 6-311+G(d,p) basis set that is available in GAMESS under WebMO. The 6-311 basis set has six Gaussian-type functions for core electrons and five Gaussian-type functions for valence electrons. The valence functions are divided into three independent groups of three, one and one. In addition, diffuse functions, which are large outer orbitals indicated by the plus sign, are included to allow electrons greater spatial freedom. Finally, polarization functions (p orbitals on H atoms, d orbitals on heavier atoms) allow improved description of bonding. CHbond.odt 3 Reagents and supplies The only chemical needed is chloroform. Each group requires enough to fill cuvettes. Supplies needed are to hold samples in the FTIR and UV-VIS-NIR spectrophotometers. • AgCl plates and holder for the FTIR. Stored in a desiccator in the lab. • 5-mm cuvette for the near-infrared (NIR) near 1700 nm. Either glass or quartz is fine. The 5-mm cuvettes are stored in the cabinet below the UV-VIS NIR instrument.. • 10-mm cuvette for the NIR near 1200 nm and 900 nm. Either glass or quartz is fine. May be in the lab. If not, check out from the stockroom. • 10-cm cuvette for the NIR near 900 and 700 nm (glass, stored below the UV-VIS NIR instrument) CHbond.odt 4 Procedure Infrared spectroscopy (include the spectrum in your report) • Start the FTIR software, if it is not already running. • Put the empty salt plates (AgCl wafers) in their holder and place that in the sample holder. Record a background spectrum from 2500 to 3500 cm-1, using a resolution of 1 cm-1. Place a drop of chloroform between the plates and record the sample spectrum. Mark the peak location. Wipe off the salt plates and return them to the desiccator. • • near-infrared spectroscopy (include all four spectra in your report) • • • • • • • Start the JASCO V-670 uv-vis-nir instrument and its software, if they are not already running. Set the NIR bandwidth to 8.0 nm. Set scanning speed to 100 nm/min and response to slow. Install the long-path cell holder, if it is not already in the instrument. ◦ To switch cell holders, first turn of the temperature control unit, if it is on. To remove the temperature-controlled cell holder, loosen the two thumbscrews at its bottom front. (The screws loosen but will not fall off.) Then gently lift the cell holder out. Set it to the side. Replace it with the long-path cell holder. Tighten the thumbscrews. Put empty 5-mm cells in the sample and reference compartments. Use the little sliding partitions to form 5-mm cell holders. Record the background spectrum from 1650-1750 nm. Place chloroform in the sample 5-mm cell and record its spectrum over the same wavelength range. The absorbance should not exceed 2.5. Adjust the baseline if necessary and locate the peak. This is the first overtone absorption. Change to empty 10-mm cuvettes, moving the sliding partitions to the appropriate places. Scan the baseline from 1100-1200 nm. Fill the sample cuvette with chloroform and record its spectrum over the same wavelength range. Adjust the baseline if necessary and locate the peak. This is the second overtone absorption. The next peak is weak so use a 100-mm (10-cm) cell. Use the 10-cm sample cell in the sample (forward) holder. Use either a second 10-cm cell in the reference (rear) holder. Change the uv-vis bandwidth to 1 nm. With an empty sample cuvette, scan the baseline from 850 to 900 nm. Fill the sample cuvette with chloroform (about 35 mL is required) and record its spectrum over the same wavelength range. Adjust the baseline if necessary and locate the peak. This is the third overtone absorption. Change the uv-vis bandwidth to 2 nm, because the next peak is weaker. Set the scanning speed to 20 nm/min. Set the data pitch to 0.2 nm. Empty the sample cuvette (no need to dry it) and scan the baseline from 710 to 730 nm. Because the peak is weak it is good to accumulate multiple spectra. Set the accumulation number (e.g., to 4) and click "accumulation" on the parameters window. Then fill the sample cuvette and record the spectrum. Adjust the baseline and locate the peak. Return your chloroform sample to the chloroform bottle. CHbond.odt 5 Calculations Spectroscopy-Based Calculations • Convert nir wavelengths to wavenumbers, cm-1. • Tabulate G(v+1)-G(0) for v=0 (fundamental), 1, 2, 3 and 4. These excitation energies, G(v+1)-G(0), are the peak locations, in cm-1. • Calculate • • Δ Gv +1/ 2 for v =0, 1, 2, 3 and 4. Examples: • ΔG1/2=G(1)-G(0) is the infrared peak location. • ΔG3/2=G(2)-G(1) = [G(2)-G(0)] - [G(1)-G(0)] is the differences between the peak locations (in cm-1) of the first overtone (NIR) and the fundamental (infrared). Graph Δ Gv +1/ 2 versus (v+1). Calculate vd and round it down to an integer. ~ Calculate ν e and D0. Convert D0 to units of kJ/mol. Use Excel's regression report ~ to get an uncertainty for ν e . • Compare D0 to a literature value (in, for example, references 1-5 and 7 and 8). • Calculate the anharmonicity constant, xe, from the slope. See equation 4. Spectroscopy-Based Uncertainty Analysis As mentioned above, regression statistics give directly an uncertainty estimate for the harmonic vibration frequency, because e equals the y intercept. We can go further, combining slope and intercept uncertainties to estimate the uncertainty of the dissociation energy. The “total differential” method of propagation of error will be used here, working from equation 6, Do = -b(b+m)/(2m). Let σb, σm and σD stand for the uncertainties in b, m and Do. The uncertainties σb and σm are results of the calculation of regression statistics. Although the symbol “σ” suggestions standard error, one could as well use a confidence interval or a multiple of the standard error. A “propagation of error” method calculates σD from σb and σm. The total-derivative method does that by considering the total differential of Do with respect to b and m. For purposes of estimating error in Do, b+m ≈ b, so Do ≈b2/(2m). (One cannot calculate Do that way, but the approximate formula is good enough for estimating uncertainty.) Then, CHbond.odt 6 ∂ Do ( ) d ( D o )= ∂b m ∂ Do ( ) db + ∂m b dm Differential changes are replaced with uncertainties, dDo→σD, db→σb, and dm→σm. Because uncertainties must be additive, they cannot cancel each other, absolute values of the partial derivatives are used. ∂ Do ∂ Do | | | | σD= ∂b σb + ∂m σm Calculate the derivatives: ∂ Do ∂b ∂ Do ∂m = 2Do b and = m b =− b2 2 m2 =− Do m . Substitute into the equation for σD. σD Do o σb + σ b m m σb σm 2 + b m | | | | = D ( | | | |) σ D =2 D b o Use the last equation to calculate the uncertainty of the dissociation energy. CHbond.odt 7 Quantum-Chemical Calculations Use the 6-311+G(d,p) basis set and density functional theory (DFT) with the B3LYP functional. The 6-311+G(d,p) basis set is the 6-311G basis set plus diffuse sp functions and one set of polarization functions on both heavy (C, Cl) and light (H) atoms. • Optimize the geometry of chloroform and the CCl3 radical. (The spin state of the radical is doublet.) Those calculations may take one hour. Record the energies. Calculate the vibration frequencies. Record the zero-point vibrational energies. Calculate the energy of the H atom (also a doublet). Because the H atom has no bonds, select the Cartesian-coordinate option in WebMO, rather than the default internal (z-matrix) coordinates. • Note which of the vibration frequencies of chloroform is the C-H stretching frequency. If in doubt, animate the vibrations, one by one. The C-H stretching ~ frequency ν e should be compared to your spectroscopy-based value. • CHbond.odt Calculate Do. Compare it to your spectroscopic result. 8 References 1. Donald F. McMillen and David M. Golden, "Hydrocarbon bond dissociation energies," Annual Reviews of Physical Chemistry, 33, 493-532, 1982. H-CCl3 is in Table 7. 2. W. Sean McGivern, Agnes Derecskei-Kovacs, Simon W. North, and Joseph S. Francisco, "Computationally efficient methodology to calculate C-H and C-X (X=F, Cl, and Br) bond dissociation energies in haloalkanes," Journal of Physical Chemistry A, 104, 436-442, 2000. 3. L. A. Gribov, I. A. Novakov, A. I. Pavlyuchko, and E. V. Vasil'ev, "Spectroscopic calculation of CH bond dissociation energy in the series of chloro derivatives of methane, ethane, and propane," Journal of Structural Chemistry, 47(4), 635-641, 2006. 4. M. L. Myrick, A. E. Greer, A. A. Nieuwland, R. J. Priore, J. Scaffidi, Danielle Andreatta, and Paula Colavita, "Birge-Sponer estimation of the C-H bond dissociation energy in chloroform using infrared, near-infrared, and visible absorption spectroscopy," Journal of Chemical Education, 85(9), 1276-1278, 2008. 5. Maia Weissman and Sidney W. Benson, "Heat of formation of the CHCl2 radical. Bond dissociation energies in chloromethanes and chloroethanes," Journal of Physical Chemistry, 87, 243-244, 1983. 6. Peter F. Bernath, Spectra of Atoms and Molecules, Oxford University Press: Oxford, 1995, page 214. 7. CRC Handbook of Physics and Chemistry, 71st ed., David R. Lide, ed., CRC Press: Boca Raton, 1990. page 9-96. 8. Darwent, B. deB., Bond Dissociation Energies in Simple Molecules, National Bureau of Standards: Washington D. C., 1970. NSRDS-NSB31. page 21. http://www.nist.gov/srd/nsrds/NSRDS-NBS31.pdf CHbond.odt 9