Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
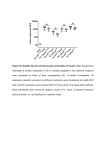
[CANCER RESEARCH 38, 1031-1035, Drug-induced April 1978] Changes in DMA Fluorescence Microfluorometry and Their Implications Intensity Detected by Flow for Analysis of DNA Content Distributions Oliver Alabaster,1 Eric Tannenbaum, Mary Cassidy Habbersett, Ian Magrath, and Chester Herman Laboratory ol Pathology ¡O.A., E. T., M. C. H., C. H.) and Pediatrie Oncology Branch ¡I.M.], National Cancer Institute, NIH, Bethesda, Maryland 20014 ABSTRACT Chicken erythrocytes, which contain less DNA than mammalian diploid cells, were used as an internal stan dard to control instrumental and staining variables during flow microfluorometric analysis. With the DNA stain, mithramycin, and with an EPICS II flow microfluorometer, ratios between the modal G, fluorescence of experimental cells and that of chicken erythrocytes were determined. The results indicate that unperturbed cell populations of L1210 and HeLa cells in vitro and L1210 ascites cells in vivo have relatively stable fluorescence ratios, although there is a significant difference between the ratios of one L1210 cell line in vitro and another in vivo. In contrast, L1210 ascites treated in vivo with different schedules of cyclophosphamide and Adriamycin showed wide fluctuations in the fluorescence intensity ratios for 96 hr after treatment. Also, differences in the fluorescence ratios were observed between less advanced and more advanced L1210 ascites after treatment with the same schedule. These effects indicate an alteration in DNA staining with mithramycin, brought about by drug treatment that could seriously affect the interpretation of DNA histogram data. Nevertheless, changes in mithramycin staining may prove to be a very important probe to detect persistent drug effects. INTRODUCTION Flow microfluorometry is used currently for rapid deter mination of relative DNA content distribution by a variety of stains that are presumed to be stoichiometric for DNA (4, 5, 11, 12, 17). These stains bind to DNA and fluoresce under appropriate light excitation in direct proportion to the DNA mass. This technique has provided relative DNA content distributions in a variety of tumor systems in perturbed and unperturbed growth conditions (21, 23, 27-30). These DNA content distributions have then been used to determine the relative position of cells in the cell cycle, and a variety of mathematical techniques have been used to derive cell cycle parameters (1-3, 14, 15, 19, 20, 22, 39). Hitherto, DNA content distributions have been obtained without the use of an internal standard. At best, serial histograms have been obtained when instrumental settings were left constant. Subsequent variations in fluorescence therefore could be partly due to instrumental or staining variables. 1 To whom requests for reprints should be addressed, at Room 1A21, Building 10, National Cancer Institute, NIH, Bethesda, Md. 20014. Received September 16, 1977; accepted January 16, 1978. APRIL Chicken erythrocytes, which have a lower DNA content than mammalian cells, fluoresce much less intensely than do normal mammalian diploid cells and therefore are well suited for use as an internal standard. The difference in fluorescence between the G, peak of mammalian cells and the G, peak of chicken erythrocytes can be used to deter mine a ratio of absolute fluorescence intensity and, hence, the DNA dry mass of the cell population (35). Furthermore, this ratio of fluorescence can be used to determine the influence of chemotherapeutic agents on the relative fluo rescence of treated cell populations. Until now it has been assumed that the DNA stoichiometry of mithramycin (and other fluorochromes) is constant even in drug-treated cell populations. However, the demonstra tion of drug-induced changes in fluorescence properties of cells must call this assumption into question. For example, if cells are not equally affected because a drug is cell cycle phase dependent, cells in protected phases of the cycle may not change their fluorescence properties as much as cells in sensitive phases of the cell cycle. Thus, under certain conditions the assumptions on the fluorescence stoichiometry of all cells in each cell cycle phase may be invalid. In the present study we have measured the fluorescent intensity of unperturbed and drug-treated cell populations and compared it to the fluorescent intensity of the internal standard. The results indicate that unperturbed cell popu lations have a reasonably stable ratio, although there is a difference between the ratio obtained from different L1210 lines in vivo and in vitro. However, in CTX2- and ADRtreated populations, there are major changes in mithramy cin staining characteristics. The possibility of drug-induced changes in stoichiometry must therefore be considered before attempting to interpret data on DNA content distributions, since assumptions re garding the relative position of all G,, S, and G2 + M cells within the DNA histogram cannot be made with any degree of certainty. However, changes in the fluorescence proper ties of mithramycin-stained, drug-treated cells could be used to estimate the duration of drug effects on both host and target tissues. MATERIALS AND METHODS Chicken erythrocytes were obtained from heparinized whole chicken blood; WBC were not removed. After centrif* The abbreviations used are: CTX, cyclophosphamide; ADR, Adriamycin; CTX/ADR, concurrent administration of both drugs, with CTX given first followed immediately by ADR. 1031 1978 Downloaded from cancerres.aacrjournals.org on August 10, 2017. © 1978 American Association for Cancer Research. O. Alabaster et al. ugation at 360 x g for 10 min, the plasma was removed, and the cells were washed with phosphate-buffered 0.85% NaCI solution, fixed in 50% ethanol, and stored at 4°.The same pool of chicken erythrocytes was used for all studies. HeLa cells grown in monolayers in Eagle's essential medium supplemented with 10% fetal calf serum were harvested daily by trypsinization [0.5% trypsin in EDTA (0.2% solution); incubated 7 to 10 min]. Growth media were changed daily. L1210 cells grown in RPMI Culture Medium 1630 (Grand Island Biological Co., Grand Island, N. Y.) were harvested during the exponential growth phase, fixed in 70% ethanol, and stored at 4°. For the in vivo studies, 5 x 105 L1210 cells i.p. of a different line, which was routinely carried in BALB/c x DBA/2 F, (hereafter called CD2F,) mice, were implanted into groups of male CD2F, mice (average weight, 21 g). On Days 5, 6, and 7 of tumor growth, 5 control mice were sacrificed by cervical dislocation, and the L1210 cells were harvested by peritoneal lavage with cold 1% sodium citrate and placed in centrifuge tubes on ice. The cells then were centrifuged at 360 x g for 5 min, the supernatant was removed, and the cell pellet was resuspended in cold 70% ethanol. All samples were stored at 4°. Cyclophosphamide (Meade Johnson Laboratory, Evansville, Ind.) was dissolved in sterile water and was Injected at a dose of 100 mg/kg i.p. Adriamycin (Adria Laboratories, Inc., Del.) was dissolved in sterile 0.85% NaCI solution and injected at a dose of 3 mg/kg i.p. Groups of CD2F! mice carrying L1210 ascites were then treated in the following 3 ways. In the first way, ADR alone was administered, and groups of 5 mice were sacrificed at 8, 16, 24, 48, 72 hr, respectively. In the second way, CTX was given first, fol lowed by ADR at 24 hr. In the third way, both drugs were given together; CTX was given first, followed immediately by ADR (CTX/ADR). Subgroups of 5 mice were then sacri ficed at 8, 16, 24, 48, 72, 96 hr, respectively. Two other groups of CD2F, mice were also treated with CTX/ADR, but the treatment differed only in that the drugs were injected 3 days after tumor inoculation in one group and 5 days after tumor inoculation in the other. Subgroups of 5 tumor-carrying mice treated on the third day postinoc ulation were then sacrificed at 24-hr intervals from Days 5 to 11; subgroups of 5 tumor-carrying mice treated 5 days postinoculation were similarly sacrificed from Days 5 to 9. Cells were harvested from the treated mice in the same way as were cells from the controls. Flow Microfluorometry. Chicken erythrocyteswere added to each cell sample so that they represented approximately 10 to 20% of the total cell number. The samples were then filtered through a 62-/¿mnylon mesh filter (Small Parts, Inc., Miami, Fla.). Mithramycin staining solution (12) at 1 x 106 cells/ml, after staining for a minimum of 20 min, was then examined under the fluorescence microscope for doublets, which never exceed 6%. Each sample was then analyzed with an EPICS II flow microfluorometer and cell sorter (Particle Technology, Inc., Los Alamos, N. M.). The princi ples of this instrument have been well described elsewhere (34). An argon ion laser at 457 nm 200 (power output, milli watts) was used to excite fluorescence. The fluorescent sig nals (at 495 nm) were then displayed on a 128-channel pulse 1032 height analyzer as a frequency distribution histogram. The photomultiplier voltage was adjusted so that the chicken erythrocytes appeared in Channel 15 (Chart 1); this ensured that the ratio was calculated well within the linear range of the amplifier. The G, peak of the chicken erythrocytes was made steady and symmetrical prior to recording the chan nel number of the G, peak of the sample. The optical alignment of the EPICS II was maintained with fluorescent microspheres (Particle Technology), which gives a coeffi cient of variation of 3.73%. When all samples had been analyzed, the ratios were calculated by dividing the cell sample G, peak channel number by the chicken erythrocyte peak channel number. The mean ratios ±S.D. were then plotted graphically; the plots are shown in Charts 2, 3, and 4. EPICS II * 4096 128 Chart 1. A typical fluorescence intensity histogram demonstrates a small chicken erythrocyte G, peak in Channel 15 (the internal standard) and the large G, peak of L1210 cells, which in this example is in Channel 72. Ordinate, cells per channel ; abscissa, increasing fluorescence intensity. The iate-S-G. + M region is off scale because the chicken erythrocytes were deliberately placed far enough upscale to be well within the linear range of the amplifier. All tumor samples were analyzed with the internal standard symmetrically placed in Channel 15 to calculate the ratio of the fluorescence of the 2 G, peaks. 1 2 3 4 5 6 ? 8 ! DAYS (Turn« Growth) Chart 2. A, fluorescence of L1210 ascites cells in vivo on Days 5, 6, and 7 of tumor growth is compared to that of chicken erythrocytes used as an internal standard. The fluorescence ratios are plotted • S.D. Cells from 2 separate cultures of L1210 cells in exponential growth, at the same cell density, have an identical fluorescence ratio, which is lower that that obtained in vivo. B, HeLa cells grown in monolayer for 9 days. The G, fluorescence ratio is compared to that of the internal standard on each day of growth. The mean ratio is 4.99 ±0.25. CANCER RESEARCH Downloaded from cancerres.aacrjournals.org on August 10, 2017. © 1978 American Association for Cancer Research. VOL. 38 Drug-Induced Changes in DNA Fluorescence W 72 96 HOURS(PostTreatment) Chart 3. A, G, fluorescence ratio of L1210 ascites cells ¡nexponential growth treated with CTX (100 mg/kg) and then with ADR (3 mg/kg) 24 hr later is compared to the ratios obtained with CTX/ADR. B, G, fluorescence ratio of L1210 ascites at the same stage of tumor growth as in A. treated with ADR (3 mg/kg) alone. The mice did not survive beyond 72 hr. Bars, ±S.D. 6 7 B 9 10 11 DAYS(PostTreatment) Chart 4. Longer term effect of CTX (100 mg/kg)/ADR (3 mg/kg) given concurrently to L1210 ascites tumor on Day 3 and Day 5 of tumor growth. No regrowth of S-phase cells was observed on Days 10 and 11 after treatment of the Day 3 tumor. S-Phase cells reappeared on Day 8 after treatment of the Day 5 tumor, and the mice did not survive beyond Day 9. Bars, ±S.D. Unstained drug-treated cells did not fluoresce when they were examined under the fluorescence microscope and in the EPICS II with the laser line at 457 nm. ratios in unperturbed cells, CTX- and ADR-treated cells showed major changes in the absolute fluorescence inten sity of the G, peak. Chart 3A demonstrates the effect of CTX alone at 8, 16, and 24 hr (open circles). At 24 hr, ADR was added so that the open circles from 48 to 96 hr represent the effect of the 2 drugs in a sequential schedule. The closed circles repre sent the effects of the 2 drugs given simultaneously. The 2 schedules produced parallel changes in the fluorescence ratio from 24 to 96 hr. When CTX was given first, however, its effect was greater and led to a persistently higher G, fluorescent intensity despite the subsequent addition of ADR. Chart 3B shows the effect of ADR alone; for the first 48 hr, the curve resembles that seen after both drugs had been given simultaneously. The effect of CTX/ADR over a longer period of time is seen in Chart 4. In this experiment the drugs were given to mice that bore L1210 ascites 3 days (closed circles) and 5 days (open circles) after inoculation of tumor, and the effects were monitored for 11 and 9 days, respectively. The Day 3 tumor-bearing mice survived more than 11 days after treatment and, at this time, have had no regrowth of S phase cells. The Day 5 tumor-bearing mice, however, had regrowth of S phase cells by Day 8 after treatment, and none survived to Day 10. Despite these differences in regrowth characteristics, the ratios were similarly depressed in both groups for the last 2 days of each study. In Chart 5, examples of histograms of different cell cycle distribution although of increased and decreased ratios are shown. In all these histograms the chicken erythrocytes are in Channel 8 so that the complete DNA content distribution of the tumor cells can be demonstrated. The Chart 5A histogram shows the cytokinetic response of the ascites tumor 16 hr after CTX was given, with a ratio of 6.25 and a large proportion of cells in the late S-G2 + M region. The Chart 56 histogram, 48 hr after CTX and ADR were given in sequence, also shows an increased ratio of 5.75, although it shows fewer S and G2 + M cells. However, the Chart 5C histogram, 96 hr after CTX/ADR treatment had been given simultaneously, also shows fewer S and Gz + M cells, but the ratio is below the control at 3.75. RESULTS The ratio of the modal G, fluorescence of the tumor cells to the modal G! fluorescence of the internal standard was calculated for each sample by histogram displays similar to that seen for untreated control cells in Chart 1. The coeffi cient of variation of the G, peak of the tumor samples ranged from 2.9 to 5.6%, whereas that of the chicken erythrocytes was 8.2 to 11.3%. In Chart 2A the mean ratio ±S.D. is plotted for 5 samples of L1210 ascites on Days 5, 6, and 7 of tumor growth. The in vitro ratio was derived from cell samples taken from 2 independent cultures of a different line of L1210 cells in exponential growth. The 2 ratios obtained from the in vitro cells were identical. Both in vivo and in vitro ratios therefore were reproducible and differed significantly. However, ¡n Chart 20 the ratio obtained from HeLa cells grown 9 days in culture showed minor fluctuations with a mean ±S.D. of 4.99 ±0.25. In contrast to the relative stability of the G, fluorescence DISCUSSION The data presented in this paper indicate that the use of chicken erythrocytes as an internal standard is a very effec tive wasy to determine relative changes in the fluorescence intensity of a cell population, with mithramycin as the DNA stain. The untreated cell populations possess relatively stable ratios (Chart 2). In contrast, the drug-treated cell popula tions show significant changes in mithramycin staining intensity, which persist for at least 11 days (Charts 3 and 4). These changes are important because of assumptions usually made about the persistence of DNA staining stoichiometry after interference with cell growth. Although the presence of a biomodal distribution of cells with a fluores cence ratio of 1:2 and an intermediate cell population suggests normal stoichiometry, the detection of a change in the fluorescence intensity of cells after drug treatment must raise the question as to whether all cells in the APRIL 1978 1033 Downloaded from cancerres.aacrjournals.org on August 10, 2017. © 1978 American Association for Cancer Research. O. Alabaster et al. EPICS II B 4*M Chart 5. Typical histograms that illustrate different ratios and cell cycle distributions. In each example the chicken erythrocytes are in Channel 8. A small peak is seen in Channel 16, which is produced by doublets. A, a histogram obtained 16 hr after a single dose of CTX. The majority of cells are in late S-G2 + M, and the ratio is increased to 6.25 (control ratio, 4.46 ± 0.21). B, a histogram obtained 48 hr after CTX and ADR were given in sequence. The majority of cells are in Q,, and the ratio is also increased at 5.25. C, a histogram obtained 96 hr after treatment with CTX/ADR. The majority of cells are also in G,, but the ratio is below the control value at 3.75. There was no evidence in any histogram that the G, fluorescence intensity ratio was influenced by the cell cycle distribution. Ordinate, cells per channel; abscissa, increasing fluorescence intensity. population have been equally affected. The G.:G, channel ratio of the control histograms had a range of 1.95 to 1.98, whereas the range in the drug-treated cell populations was 1.83 to 2.13. The wider variability of the G2:Gi ratio in the drug-treated group therefore supports the possibility of an unequal cell response. As can be seen from Chart 3A, administration of CTX alone raises the fluorescence ratio substantially at 8, 16, and 24 hr. CTX, which is an alkylating agent thought to affect cells in all phases of the cell cycle (10, 26, 37), is known to induce cross linkages in DNA either by linking adjacent areas of 1 strand or by joining together opposite strands of the helix (7). Furthermore, alkylating agents can 1034 induce not only development of giant cells with increased amounts of DNA and protein (8, 9) but also chromosomal aberrations (32, 33, 36). However, ADR binds to DNA, and this could influence the availability of mithramycin-binding sites. From Chart 3A it is evident that simultaneous treatment with ADR limits the increased fluorescence induced by CTX, so that the effects more closely resemble those seen with ADR alone (Chart 3B). The changes seen in both groups from 48 to 96 hr (Chart 3A) are very difficult to explain simply in terms of drug binding because fluorescence both increases and de creases in relation to the controls after treatment. There fore, it seems more likely that these changes in fluores cence are due to structural alteration of the chromatin. Furthermore, nothing is known of the precise metabolic interaction of CTX and ADR, which might explain these observations. An increase in the fluorescence ratio in unperturbed cell populations is consistent with an increase in absolute DNA dry mass (35). However, in drug-treated populations, the greatest increase in the fluorescence ratio is within 8 hr, and this is therefore probably not due to a true increase in DNA mass. Also, it is probably not due to quantitative changes in mitochondrial DNA since this represents such a small proportion of the total cellular DNA. The subsequent fall in the fluorescence ratio (Charts 3 and 4) below the control value is also probably not due to changes in DNA mass because subsequent increases in DNA mass would have to have occurred so rapidly. This is evident in Chart 3/4 from 72 to 96 hr. Interpretation of all these observations is further compli cated by the data seen in Chart 4. These data indicate a different pattern of response in L1210 tumor cell popula tions treated at different intervals after implantation. Not only is there a difference in the fluorescence ratios, but also there is a difference in the regrowth characteristics. The more advanced tumor resumed cell growth (reappearance of S-phase cells) on Day 8, whereas the less advanced tumor had no evidence of regrowth by Day 11. However, the fluorescence ratios in the advanced tumor on Days 8 and 9 were very similar to those seen on Days 10 and 11 in the less advanced tumor. Therefore, the fluorescence changes appear to be independent of cell proliferation and dependent on other factors. The data presented in Chart 4 indicate that drug effects may persist for some time. It is known that both CTX and ADR may cause long-term chromosomal damage and are capable of inducing malignancy (6, 16, 18, 25, 33, 35). Thus, it is possible that persistent changes in fluorescence ratios might be a way to detect permanent drug effects on host tissues as well as the persistence of drug effects on tumor cell populations. Finally, what is of most immmediate significance is that changes in cell fluorescence may effect the interpretation of DNA histograms. Not only may some drugs have selective effects on cells in different phases of the cell cycle in lethality or arrest of cell cycle progression, but also these effects may be absent or reduced in resistant subpopulations of cells. More sensitive cells might then change their fluorescence properties to a greater extent than would less CANCER RESEARCH Downloaded from cancerres.aacrjournals.org on August 10, 2017. © 1978 American Association for Cancer Research. VOL. 38 Drug-Induced Changes in DNA Fluorescence 15. Dethlefsen, L. A., Gray, J. M., George, Y. S., and Johnson, S. Flow Cytometric Analysis of the Perturbed Cellular Kinetics of Solid Tumors: Problems and Promises. In: W. Go'hde, J. Schumann, and T. H. Büchner (eds.), Second International Symposium on Pulse-Cytophotometry, pp. 188-200. Ghent, Belgium: European Press Medikon, 1976. 16. DiPaolo, J. A. Teratogenic Agents: Mammalian Test Systems and Chem icals. Ann. N. Y. Acad. Sci., Õ63:801-812, 1969. 17. Dittrich, W., and Göhde,W. Impulse Cytophotometry of Single Cells in Suspension. Z. Naturforsch., 24b: 360-361, 1969. 18. Fahmy, 0. G., and Fahmy, M. The Genetic Effects of the Biological Alkylating Agents with Reference to the Pesticides. Ann. N. Y. Acad. Sci., 160: 228-243, 1969. 19. Fried, J. Method for Quantitative Evaluation of Data from Flow Micro fluorometry. Computers Biomed. Res., 9: 263-276, 1976. 20. Fried, J., Yataganas, X., Kitahara, T., Perez, A., Ferguson, R., Sullivan, S., and Clarkson, B. Quantitative Analysis of Flow Microfluorometric Data from a Synchronous and Drug Treated Cell Populations. Computer Biomed. Res., 9: 277-290, 1976. ACKNOWLEDGMENTS 21. Göhde,W., J. Schumann, and T. H. Büchner(eds.). Second Interna tional Symposium on Pulse-Cytophotometry. Ghent, Belgium: European The authors express their appreciation to Dr. Kwang Woo for the cultured Press Medikon, 1976. Hela cells, Donna Roberts for her secretarial assistance, and Ralph Isenberg 22. Gray, J. W. Cell Cycle Analysis from Computer Synthesis of Deoxyribo for his photographic services. nucleic Acid Histograms. J. Histochem. Cytochem., 22: 642-650, 1974. 23. C. A. M. Haanen, H. F. P. Hillen, and J. M. C. Wessels (eds.). First International Symposium on Pulse-Cytophotometry. Ghent, Belgium: European Press Medikon, 1975. REFERENCES 24. Liedeman, R., and Bolund, L. Acridine Orange Binding to Chromatin of Individual Cells and Nuclei under Different Staining Conditions. I and II. 1. Alabaster, 0., and Bunnag, B. Flow Microfluorimetric Analysis of Sensi Exptl. Cell Res., 101: 164-183, 1976. tive and Resistant Leukemia L1210 following 1-/3-Arabinofuranosylcytosine in Vivo. Cancer Res., 36. 2744-2749, 1976. 25. Loveless, A. Genetic and Allied Effects of Alkylating Agents. London: 2. Baisch, H., Göhde,W., and Linden, W. A. Mathematical Analysis of ICPButterworth & Co., 1966. 26. Lundlum, D. B. Molecular Biology of Alkylation: An Overview. In: A. C. Data to Determine the Fraction of Cells in the Various Phases of Cell Sartorelli and D. G. Johns (eds.), Antineoplastic and Immunosuppressive Cycle. In: C. A. M. Haanen, H. F. P. Hillen, and J. M. C. Wessels, (eds.), Agents, Part 2, Chap. 31, pp. 6-17. Berlin: Springer-Verlag, 1975. First International Symposium on Pulse-Cytophotometry, pp. 68-76. 27. D. Lutz (ed.), Third International Symposium on Pulse-Cytophotometry. Ghent, Belgium: European Press Medikon, 1975. 3. Baisch, H., Linden, W. A. Different Mathematical Models for PulseGhent, Belgium: European Press Medikon, in press. 28. Mayall,, B. H. (ed.). Papers from the Third Engineering Foundation Cytophotometric Evaluations Applied to Asynchronous and Partially Conference on Automated Cytology. J. Histochem. Cytochem., 22: 451Synchronised Cell Populations. In: C. A. M. Haanen, H. F. P. Hillen, and J. M. C. Wessels (eds.), First International Symposium on Pulse-Cyto 765. photometry, pp. 61-67. Ghent, Belgium: European Press Medikon, 1975. 29. Mayall, B. H. (ed.). Papers from the Fourth Engineering Foundation Conference on Automated Cytology. J. Histochem. Cytochem., 24: 14. Barlogie, B., Zante, J., Drewinko, B., Schumann, J., Búchner, T., Göhde,W., Hart, J. S., and Johnston, D. A. The Use of Mithramycin in 414,1976. Pulse-Cytophotometry. In: W. Göhde,J. Schumann, and T. H. Büchner 30. Mayall, B. H., and Gledhill, b. L. (eds.). Papers from the Fifth Engineer (eds.), Second International Symposium on Pulse-Cytophotometry, pp. ing Foundation Conference on Automated Cytology. J. Histochem. 125-136. Ghent, Belgium: European Press Medikon, 1976. Cytochem., 25: 479-952, 1977. 5. Berhan, E. Pulse-Cytophotometry as a Method for Rapid Photometer 31. Nicolini, C., and Baserga, R. Circular Dichroism Spectra and Ethidium Bromide binding of 5-Deoxybromouridine-substituted Chromatin. Bio Analysis of Cells. In: C. A. M. Haanen, H. F. P. Hillen, and J. M. C. Wessels (eds.), First International Symposium on Pulse-Cytophotometry, chem., Biophys. Res. Commun., 64: 189-195, 1975. pp. 15-21. Ghent, Belgium: European Press Medikon, 1975. 32. Ochoa, M., Jr., and Hirschberg, E. Alkylating Agents. In: R. J. Schnitzer and F. Hawking (eds.), Experimental Chemotherapy, Vol. 5, pp. 1-132. 6. Brookes, P., and Lawley, P. D. The Reaction of Mustard Gas with Nucleic Acids in Vivo and in Vitro. Biochem. J., 77: 478-484, 1960. New York: Academic Press, Inc., 1967. 7. Brookes, P., and Lawley, P. D. The Reaction of Mono and Di-Functional 33. Sieber, S. M., and Adamson, R. M. The Clastogenic, Mutagenic, Tera Alkylating Agents with Nucleic Acids. Biochem. J., 80: 496-503, 1961. togenic, and Carcinogenic Effects of Various Antineoplastic Agents. In: 8. Caspersson, T., Farber, S., Foley, G. E., and Killander, D. Cytochemical Pharmacological Basis of Cancer Chemotherapy, The University of Observations on the Nucleolus-Ribosome System. Effects of ActinomyTexas System Cancer Center M. D. Anderson Hospital and Tumor cin D and Nitrogen Mustard. Exptl. Cell Res.. 32: 528-552, 1963. Institute, pp. 401-468. Baltimore: Williams & Wilkins, 1975. 9. Cohen, L. S., and Studzinski, G. P. Correlation between Cell Enlarge 34. Steinkamp, J. S., Fulwyler, M. J., Coulter, J. R., Hiebert, R. D., Horney, ment and Nucleic Acid and Protein Content of HeLa Cells in Unbalanced J. L., and Mullaney. P. F. A New Multiparameter Separator of Micro scopic Particles and Biological Cells. Rev. Sci. Instr., 44: 1301-1310, Growth Produced by Inhibition of DNA Synthesis. J. Cellular Physiol., 69: 331-339, 1967. 1973. 10. Connors, T. A. Mechanism of Action of 2-Chloroethylamine Derivatives, 35. Tannenbaum, E., Cassidy Habbersett, M., Alabaster, O., and Herman, C. Sulfur Mustards, Epoxides, and Aziridines. In: A. C. Sartorelli and D. G. J. Measurement of Cellular DNA Mass by Flow Microfluorometry Using a Johns (eds.), Immunosuppressive and Antineoplastic Agents, Vol. 2, Biological Internal Standard. J. Histochem. Cytochem., in press, 1978. Chap. 32, pp. 18-34. Berlin: Springer-Verlag, 1975. 36. Vogel, F., and Roehrborn, G. (eds.). Chemical Mutagenesis in Mammals and Man Berlin:Springer-Verlag, 1970. 11. Crissrnan, H. A., Oka, M. S. and Steinkamp, J. A. Rapid Staining Methods for Analysis of Deoxyribonucleic Acid and Protein in Mamma 37. Wheeler, G. P. Studies Related to the Mechanisms of Action of Cytotoxic lian Cells. J. Histochem. Cytochem, 24: 64-71, 1976. Alkylating Agents: A Review. Cancer Res., 22: 651-688, 1962. 12. Crissrnan, H. A., and Tobey, R. A. Cell Cycle Analysis in 20 Minutes. 38. Wheeler, G. P. Alkylating Agents. In: J. F. Holland and E. Frei, III, (eds.), Science, 184: 1297-1298, 1974. Cancer Medicine, Chap. XIII-4, pp. 791-806, Philadelphia: Lea & Febiger, 13. Darzynkiewicz, Z., Tráganos, F., Sharpless, T. K., and Melamed, M. R. 1973. Cell Cycle-related Changes in Nuclear Chromatin of Stimulated Lympho 39. Woo, K. B. The Discrete-Time Kinetic Model Analysis of DNA Content cytes as Measured by Flow Cytometry. Cancer Res., 37: 4635-4640, Distributions in Experimental Tumor Cells. Cell Tissue Kinet , in press 1977. 1978. 14. Dean, P. N., and Jett, J. H. Mathematical Analysis of DNA Distributions 40. Yonuschot, G., and Muschrush, G. W.Terbium as a Fluorescent Probe Derived from Flow Microfluorometry. J. Cell Biol., 60: 523-527, 1974. for DNA and Chromatin. Biochemistry 14: 1667-1681, 1975. sensitive cells. Hypothetically, some G, cells could appear within theG2 peak or wee versa. Under these circumstances the detection and interpretation of cell cycle parameters would be very difficult. Further studies are required with well-established tech niques (13, 24, 31, 40) used to assess whether these are really changes in chromatin structure. It will also be neces sary to examine other DNA dyes to determine whether these fluorescence changes are a common finding or whether mithramycin is a unique probe that could be used to detect persistent drug effects and cell damage. APRIL 1978 Downloaded from cancerres.aacrjournals.org on August 10, 2017. © 1978 American Association for Cancer Research. 1035 Drug-induced Changes in DNA Fluorescence Intensity Detected by Flow Microfluorometry and Their Implications for Analysis of DNA Content Distributions Oliver Alabaster, Eric Tannenbaum, Mary Cassidy Habbersett, et al. Cancer Res 1978;38:1031-1035. Updated version E-mail alerts Reprints and Subscriptions Permissions Access the most recent version of this article at: http://cancerres.aacrjournals.org/content/38/4/1031 Sign up to receive free email-alerts related to this article or journal. To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at [email protected]. To request permission to re-use all or part of this article, contact the AACR Publications Department at [email protected]. Downloaded from cancerres.aacrjournals.org on August 10, 2017. © 1978 American Association for Cancer Research.