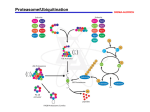
Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Send Orders of Reprints at [email protected] Current Pharmaceutical Design, 2013, 19, 000-000 1 Emerging Role of the Ubiquitin-proteasome System as Drug Targets Gozde Kar1, Ozlem Keskin1, Franca Fraternali2 and Attila Gursoy1,* 1 Center for Computational Biology and Bioinformatics and College of Engineering, Koc University, Rumelifeneri Yolu, 34450 Sariyer Istanbul, Turkey; 2Randall Division of Cell and Molecular Biophysics, New Hunt's House, King's College London, Guy's Campus, SE1 1UL London UK Abstract: The ubiquitin-proteosome system (UPS) regulates a wide range of cellular processes including protein degradation, DNA repair, transcription regulation, and cell signaling. Alterations and mutations in UPS components give rise to various human diseases, most prominently cancer and neurodegenerative disorders. Here, we review recent advances in UPS-based drug discovery highlighting the emerging relationships between the UPS and disease and discuss potential future therapeutic interventions. In particular, we focus on recent structural approaches in UPS and explain how the knowledge of protein structural details can guide the design of specifically targeted inhibitors. Keywords: Ubiquitin-proteosome system, disease, drug design. 1. INTRODUCTION The ubiquitin-proteosome system (UPS) is essential for the protein turnover and has a wide variety of functions ranging from cell-cycle control and transcription to development [1]. Ubiquitin is a small 76-aminoacid protein that can reversibly bind to and label other proteins in a highly specific manner. Ubiquitin attachment takes place via a sequential enzymatic route involving ubiquitin activation (by E1 enzymes), ubiquitin conjugation (by E2 enzymes), and ubiquitin substrate tagging (by E3 enzymes). In the first step, ubiquitin is activated by an ATP-dependent E1 and E1 delivers the activated ubiquitin to E2 through a transthiolation reaction. E3s recognize specific target substrates and catalyze the ubiquitin transfer from the E2 to the substrate. Ubiquitin can be conjugated to the substrate as a monomer on one (monoubiquitination) or more substrate lysines (multiubiquitination) or as a polymer (polyubiquitination) by forming ubiquitin chains [2]. Seven lysine residues of ubiquitin can participate in ubiquitin conjugation, leading to ubiquitin chains of different lengths, topologies and functions. Substrates that are tagged by ubiquitin chains conjugated through the lysine 48 of ubiquitin are targeted by proteosome and undergo degradation process, whereas chains linked at lysine 63 are associated with signaling and trafficking events [3]. Importantly, ubiquitination is a reversible process; the deubiquitinating enzymes (DUBs) can in fact remove ubiquitins from modified substrates, thus rescuing the substrate from degradation. Considering the crucial multiple biological roles of UPS enzymes, it is expected that malfunctioning of any component participating in the regulation of these enzymes would result in disease. There is in fact accumulating evidence of altered functions of UPS enzymes in numerous cancer types [4], cardiovascular and viral diseases [5, 6] and neurodegenerative disorders [7]. In treating such diseases, developing efficient small-molecule inhibitors targeting the UPS pathway rather than the single molecules is of crucial importance and has elicited significant interest in recent drug design studies. In this review, we first summarize role of UPS in human diseases along with the current approaches in the development of small-molecule inhibitors targeting UPS components. We then discuss the potential future therapeutic interventions, focusing *Address correspondence to this author at the Center for Computational Biology and Bioinformatics and College of Engineering, Koc University, Rumelifeneri Yolu, 34450 Sariyer Istanbul, Turkey; Tel:/Fax: ???????????????????????; E-mail: [email protected] 1381-6128/13 $58.00+.00 particularly on targeting protein-protein interactions and interfaces of UPS components in disease with the ultimate purpose of designing more efficient and selective inhibitors. 2. TARGETING UPS IN DISEASE Ubiquitin attachment to proteins (ubiquitination) is a major post-translational modification that can have profound effects on protein stability, localization or interaction pattern [8]. Several proteins regulated by ubiquitination control cell signaling events, and hence are crucial for cell-cycle progression, proliferation and apoptosis [8]. Such signaling processes are frequently altered in cancer with several tumor suppressors and oncogenes representing the enzymes of the ubiquitin conjugation and deconjugation pathways. The first inhibitor targeting the proteosomal degradation of proteins in cancer is bortezomib, which has illustrated a clinical success and stimulated further research in the design of specific UPS-based inhibitors. Subsequently, other UPS enzymes such as E1, E2 and E3 have started to be exploited as targets in disease. Below, we start by describing the role of each enzyme classes (E1, E2, E3, DUBs and proteasome) in the UPS pathway and in disease and by listing all the so far available inhibitors and therapeutic approaches. The UPS pathway and the various possibilities for therapeutic intervention are displayed in (Fig. 1). Targeting E1 Activating Enzymes The first step of the UPS pathway is ubiquitin activation, performed by E1 enzymes. Ubiquitin (Ub) or ubiquitin-like proteins (Ubl) are adenylated at their C-terminal glycine residue and react with the E1 active site thiol to become charged as a thiol ester. Then, Ub or Ubl are transferred to E2 conjugating enzymes also through a thiol ester bond. There are eight structurally and functionally similar E1s that can activate Ub and Ubls. Although Ubactivating E1s can charge a variety of E2s, Ubl-activating E1s appear to charge only a single E2 [1]. For inhibiting ubiquitin activation, there are a couple of points to target (Fig. 1). One of them is to prevent ATP from binding to E1 and hence to inhibit Ub-adenylate formation using small-molecule inhibitors. Another possibility is to target the E1 active site thiol for blocking the formation of E1-Ub complex. One example is PYR41 which is a pyrazone derivative designed for inhibiting HDM2dependent p53 ubiquitination. The nitrogen dioxide group of PYR41 covalently modifies the active site cysteine of E1 while, notably, it does not inhibit other thiol-dependent enzymes including many E2s [9]. The downstream effects of PYR41 treatment are NF© 2013 Bentham Science Publishers 2 Current Pharmaceutical Design, 2013, Vol. 19, No. 00 Kar et al. Fig. (1). UPS mechanism and several potential UPS-based drug targets. Ubiquitin attachment takes place via a sequential enzymatic route. In the first step, ubiquitin is activated by an ATP-dependent E1 and E1 delivers the activated ubiquitin to E2 through a transthiolation reaction. E3s recognize specific target substrates and catalyze the ubiquitin transfer from the E2 to the substrate. Substrates that are tagged by ubiquitin chains conjugated through the lysine 48 of ubiquitin are targeted by proteosome and undergo degradation process, whereas monoubiquitination is associated with signaling and localization events [3]. The deubiquitinating enzymes (DUBs) can remove ubiquitins from modified substrates. There are several potential drug targets in UPS in disease: the activity of E1, E2, E3 enzymes, DUB and proteasome can be inhibited. Additionally, inhibition by blocking the interface regions of E1-E2, E2-E3 and E3-susbtrate interactions would provide an efficient means in treating UPS-associated diseases. B activation, stabilization of p53 and induction of p53-dependent transcription [9]. Besides PYR41, the nitric oxide (NO)-producing drug JS-K also inhibits E1-ubiquitin thioester formation through an interaction between the E1 active-site thiol and NO [10]. Similarly, this compound results in an increased p53 expression and decreased levels of total ubiquitinated proteins. E1s can also be inhibited through adduct formation. One example of such compounds is Nedd8-activating enzyme (NAE-E1) inhibitor; MLN4924, which is an adenosine sulphamate analogue [11, 12] that binds to Nedd8 thioster form of NAE-E1 and then forms a covalent adduct with Nedd8. Blocking NAE by MLN4924 results in the inhibition of CRL (Cullin-Ring Finger Ligase) activity and thus stabilization of CRL substrates (such as CDT1), leading to deregulation of DNA synthesis during the S phase of the cell division cycle and apoptosis in proliferating cells [11]. MLN4924 has entered Phase II clinical trials for treating multiple myeloma and non-Hodgkin’s lymphoma. (~500 or more have been proposed to exist in human) [18], which suggests that one E2 can recognize several different E3s [19]. The principles determining the E2-E3 selectivity are unclear and in many cases it is not known which E3s interact with which E2s [20]. Since the knowledge about the functions of E2s is also limited, it is difficult to design selective E2 inhibitors. Despite these challenges, there are emerging approaches for inhibiting E2s (Fig. 1). One example for blocking E1-E2 interaction is a synthetic peptide called UBC12N26, which was shown to compete for UBC12-NAE binding, hence blocking transthiolation of Nedd8 to UBC12 and preventing the transfer of Nedd8 to the substrate targets [21]. For some E2s such as UBE2G2, E3 binding can allosterically activate ubiquitination activity, then another potential mode of inhibition may be the blocking E2-dependent ubiquitin transfer using allosteric effectors. An additional approach is to inhibit catalytic activity of E2s by targeting the conserved asparagine residue of E2s. Targeting E2 Conjugating Enzymes E2s are involved in ubiquitin conjugation and ubiquitin transfer to the substrates. E2s mediate ubiquitin chain assembly and ubiquitin linkage topology, therefore together with E3s, leading to determination of the fate of the substrates. E2s have been implicated in many diseases including cancer. For example, UBCH10 was observed in chromosomal instability [13], UBE2T in lung cancer [14], UBE2Q2 in head and neck squamous cell carcinoma [15] and the SUMO E2 UBC9 in ovarian carcinoma and breast cancer [16, 17]. Currently, there is a limited number of known E2s (~40 in human); however the number of known E3s is increasing rapidly Targeting E3 Ubiquitin Ligases E3s recognize specific target substrates and catalyze the ubiquitin transfer from the E2 to the substrate. E3s are represented by three main classes: Homologs of E6AP Carboxy Terminus (HECT), Really Interesting New Gene (RING), and the UFD2 homology (Ubox) family proteins. HECT is a domain of ~350 amino acids; its N-terminal lobe contains the E2-binding site, and the C-terminal lobe confers catalytic activity. The conserved Cys residue in the Cterminal lobe forms thioester complexes with ubiquitin [22]. The RING finger domain contains a short motif rich in histidine and cysteine residues that coordinate zinc atoms in a cross-brace structure, characterized by a central -helix and variable-length loops separated by small beta strands [23]. The U-box domain is similar Emerging Role of the Ubiquitin-proteasome System to the structure of the RING domain with the exception that it lacks the conserved histidine and cysteine residues [24]. The ubiquitination mechanisms for these three E3 classes differ. For HECT domain E3s, the ubiquitin is first transferred from E2 to the active site residue of HECT E3, with subsequent transfer from E3 to the substrate protein. For RING and U-box type E3s, ubiquitin is directly transferred from E2 to the substrate without an E3 intermediate linkage. Although the human genome encodes numerous E3 ligases [25] and there are several efforts in identifying functions of E3s and their association to human disease, progresses in this field are still in its infancy. In the UPS, E3 ligases are probably the most attractive drug targets since the substrate selectivity relies mainly on the specificity of E3s. Potential points to target are: E2-E3 binding interface, E3-substrate recognition and the activity of the E3 itself (Fig. 1). Below, we present E3s, their alterations in diseases, the available inhibitors targeting these E3s; a summary is provided in Table 1. RING-finger Domain Type E3s Mdm2 is a RING-finger E3 ligase that mediates ubiquitination of tumor suppressor protein p53. Mdm2 is an oncogenic protein whose gene amplification and overexpression are prominently present in human cancers including breast cancer, lung cancers and many other tumor types [26, 27]. There have been so far a numerous approaches that have proposed compounds disrupting the interaction between E3 and its substrates. One of such inhibitors is Nutlin /R7112, which has entered clinical trials. Nutlin /R7112 targets the interface of E3 ligase MDM2 (HDM2) and its substrate p53 to prevent p53 degradation and thus to elevate p53 expression [28]. Similarly, the compound RITA is used to promote p53 stabilization [29]. Known disease associations include various cancers; breast and lung, blood cancers and multiple myeloma [30]. Another ligase class that can be targeted in multiple cancers and disorders linked to the NF-B pathway is cullin-RING E3 ligases (CRLs). SCFSkp2 is a CRL that ubiquitinates p27 targeting it for proteosomal degradation [31]. Elevated levels of SCFSkp2 and decreased levels of p27 are observed in some human cancers. Chen et al. [32] have identified a compound that is active against leukemia cells and prevent the formation of the functional SCF complex, which stabilizes p27, induce G1/S cell cycle arrest, and thereby triggers cell death [32]. SCFTRCP is a CRL that mediates the degradation of IB; the inhibitory component of the proinflammatory transcription factor NF-B [33]. An inhibitor of SCFTRCP was found as well, which prevents the polyubiquitination and subsequent degradation of IB [33]. Another example is the inhibitor of Cdc4, the yeast ortholog of the mammalian Cullin RING E3 ligase Fbw7, that was identified in a biochemical screen and disrupts binding of Cdc4 to its substrate SIC1 [34]. Inhibition is achieved through an allosteric mechanism: the inhibitor binds to Cdc4 at a site distant from SIC1 binding, which induces a large conformational change resulting in a distortion of the substrate binding pocket and impediment of the substrate recognition [34]. Although these inhibitors of CRL have not entered into clinical trials yet, they represent a strong new avenue for treating several human diseases. The Inhibitors of Apoptosis Proteins (IAPs) are also actively targeted E3 ligases in disease. They mediate degradation of several substrates involved in apoptosis and signaling and are linked to several cancer types such as liver and lung, ovarian carcinoma, solid and lymphoid tumors [30]. There are a few antagonists of IAPS such as GDC-0152, LCL161, YM155 and TL32711 that have entered clinical trials. These compounds are mimetics of SMAC proteins that antagonize IAPs and promote IAP auto-ubiquitination and degradation, leading to the death of cancerous cells through TNF pathway [35]. BRCA1 (Breast cancer type 1 susceptibility protein) is an E3 ligase with tumor suppressor activity that is involved in DNA dam- Current Pharmaceutical Design, 2013, Vol. 19, No. 00 3 age repair and cell cycle check point control processes. It is mutated in more than 50% of inherited breast cancers [36] and its mutation leads to early-onset breast and ovarian cancers and to the development of a malignant phenotype [37]. BRCA1 heterodimerizes with another RING finger protein, BRCA1 Associated Ring Domain 1 (BARD1) increasing its ubiquitin ligase activity [38]. The Parkin gene encodes a RING finger E3 that can ubiquitinate itself and the -synuclein interacting protein, Synphilin 1. Familial associated mutations in Parkin leads to a deficiency in E3 activity and thus to Parkinson’s disease [39]. Histopathological hallmark of Parkinson’s disease are the so-called Lewy bodies which are abnormal protein aggregates developing inside nerve cells. Lew bodies contain ubiquitin and filamentous aggregates of denatured proteins including -synuclein [40]. Another E3 ubiquitin ligase is VHL (von Hippel-Lindau) that is mutated in a familial cancer susceptibility syndrome, von HippelLindau syndrome. This disease is characterized by the development of various highly angiogenic tumors such as renal cell carcinoma, pancreatic cysts and tumors [41]. VHL ligase functions as a substrate recognition subunit in the VCB-Cullin 2-VHL complex and mediates ubiquitination of HIF-1 (Hypoxia-inducible factor-1) protein [42]. HIF-1 functions in oxygen homeostasis and is a regulator of energy metabolism and angiogenesis processes [42]. Under normal oxygen levels, HIF-1 is hydroxylated, recognized by VHL and ubiquitinated by VCB-Cullin 2-VHL complex leading to its proteasomal degradation. However, hypoxia decreases the capacity of prolyl hydroxylases to hydroxylate HIF-1, and in turn HIF-1 escapes from VHL-induced degradation, which promotes expression of hypoxia inducible genes such as Glucose Transporter 1 and Vascular Endothelial Growth Factor (VEGF) [4]. Mutations in VHL are observed to prevent degradation of HIF-1 which leads to upregulation of HIF-1-induced genes and to high levels of tumor vascularization [43]. Consequently, reactivating the VHL ligase would be a possible strategy to treat VHL-associated tumors. In one such effort, a bioengineered VHL complex is exploited to increase HIF-1 degradation [44]. HECT Domain Type E3s E6-Associated Protein (E6-AP) is a member of HECT E3 ubiquitin ligase family that catalyzes ubiquitination and subsequent degradation of p53 upon infection by Highrisk human papillomaviruses (HPV) [45]. HPV 16 and 18 are associated prominently with cervical cancer together with genital and head and neck cancers [46]. Additionally, mutations in E6-AP are associated with Angelman’s syndrome [47]. ARF-BP1 (HUWE1) is another HECT-type E3 ligase that can induce p53 ubiquitination and degradation [48]. ARF-BP1 is highly expressed in breast cancer cell lines and therefore appears to have a potential role in breast cancer tumorigenesis [49]. Another HECT type E3 is Nedd4-1, belonging to Nedd4-like subgroup, negatively regulates tumor suppressor Phosphatase and Tensin Homolog protein (PTEN) by ubiquitinating it for degradation [50]. Overexpression of Nedd4-1 was detected in multiple human cancer samples and the depletion of Nedd4-1 inhibited xenograft tumor growth in a PTEN-dependent manner [50]. On the contrary, one study reported no dysregulation of PTEN protein level upon Nedd4-1 knockout [51]. PTEN is frequently mutated in human tumors [52]. A more exact explanation of the role of Nedd41 in regulating PTEN is therefore necessary to elucidate mechanisms of tumor formation and progression. Smurf2 is also a HECT E3 and has a key function in mediating TGF signaling pathway [53, 54]. It mediates ubiquitination of Smad1, Smad2 and TGF receptor 1. High-level expression of Smurf2 has been observed to correlate with tumor development, lymph node metastasis and a poor survival rate in esophageal squamous cell cancer [55]. 4 Current Pharmaceutical Design, 2013, Vol. 19, No. 00 Table 1. Kar et al. E3 ligases and Disease E3 Substrate Associated disease Alterations in disease Inhibitor/Treatment Brca1 NPM1 [170], RBBP8 [171], RNA polymerase [172], ESR1 [173] Breast and ovarian cancer [37] Promoter hypermethylation [174], genomic deletions and duplications [175] - Cervical, genital, head and neck carcinomas [45, 177], Angelman’s syndrome [47] Mutations associated with Angelman’s syndrome [47], infection by high-risk HPV in cancer [45, 177] mRNA decay factor TTP promotes rapid decay of E6-AP and stabilizes p53 in cervical cancer [178] (RING-finger) E6-AP p53 [45], (HECT) p27 [176] IAP Several substrates Various cancers including liver, lung, ovarian carcinoma, lymphoma Overexpression of IAPs in various cancers [179] Many inhibitors targeting IAPs such as GDC-0152, AEG35156, YM155, LCL161, AT-406 p53 and p27 [180] Various cancers including breast and lung Overexpression and amplification [27], point mutations and nucleotide insertions in human tumors [181] Nutlins, RITA, Parthenolide PTEN [50] Multiple cancers, including bladder, prostate, colorectal and gastric cancer [50, 182] Overexpression and mutations in cancer [50, 182] - PARKIN, Synphilin 1 [39] Parkinson’s disease [39] Familial associated mutations [39] (RING-finger) Nitric oxide to inhibit Parkin’s ligase activity [183] SCFTRCP IB [184] Multiple cancers and NF-B related disorders Overexpression Inhibitor to prevent SCFTRCP activity [33] (RING-finger) Notch [185], c-Myc [186], c-Jun [187], cyclin E [188] and mTOR [189] Colon, stomach cancer, T-cell acute lymphocytic leukemia [190] Mutations in substrate recognition domain [190] Allosteric inhibitor of its yeast ortholog, Cdc4 impedes recognition of the substrate [34] SCFSkp2 p27 [191] Multiple cancers including breast and colon [192], gastric, lung, prostate, ovarian cancer [191] Gene amplification and overexpression in cancer [193] Inhibitor to prevent SCFSkp2 activity and stabilize p27 [32] Esophageal squamous cell cancer [55] Overexpression [55] - (HECT) Smad1 [53], Smad2 [54], TGF receptor 1 [194] VHL HIF-1 [42] Renal cell carcinoma, pancreatic cancer, VHL syndrome [195] Germline deletion, mutations [43] Bioengineered VHL complex to increase HIF-1 degradation [44] p53 [57], p63 [196], KLF5 [197], ERBB4 [198] Breast and prostate cancer [56] Overexpression [56] - MEKK2, AIF, TGF-activated kinase [199, 200] Breast cancer and acute myeloid leukaemia [199, 201] Overexpression [202] Inhibitor compound AEG35156 to inhibit XIAP [203] (RING-finger) Mdm2 (RING-finger) Nedd4-1 (HECT) Parkin (RING-finger) SCFFbw7 (RING-finger) Smurf2 (RING-finger) Wwp1 (HECT) Xiap (RING-finger) E3 ligases together with their substrates, associated disease, alterations and available inhibitors/treatment are listed. Wwp1 is a HECT E3 that is aberrantly regulated in cancer. Overexpression of Wwp1 is observed in breast and prostate cancer samples [56]. Wwp1 has been shown to regulate p53 ubiquitination tanslocating p53 to the cytoplasm and diminishing its transcriptional activity [57]. Inactivation of Wwp1, therefore, could activate p53, which may provide a means for treating cancer. The HECT E3 ligases may be more amenable to smallmolecule intervention than other classes of E3s as they form cova- lent thioester intermediates with ubiquitin before transferring ubiquitin to the substrate. Since this step requires a large conformational change in the HECT domain, it may be possible to block this transition by small-molecule inhibitors [1]. Targeting Deubiquitinating Enzymes Deubiquitinating enzymes regulate various ubiquitin-mediated biological processes such as proteosome-dependent degradation, Emerging Role of the Ubiquitin-proteasome System Current Pharmaceutical Design, 2013, Vol. 19, No. 00 protein localization and endocytosis by reversing the action of the ubiquitin conjugation mechanism [58]. Deubiquitinases belong to five different gene families; four are cysteine peptidases (the ubiquitin-specific proteases (USPs), the ubiquitin C-terminal hydrolases (UCHs), ovarian tumor domain proteins (OTU), and MachadoJoseph disease protein (MJD), and the fifth family consists of metalloproteinases from the JAMM/MPN domain family [58]. The dysregulation of DUBs is highly observed in human diseases, especially cancer. In recent years, there is a growing interest in developing inhibitors against DUBs although there has been a limited success in turning these into drugs [59]. The DUBs have been extensively reviewed in [59]. Here, we classify the DUBs according to their domain type, overview advances in targeting these DUBs and provide a summary in Table 2. USP Family of Deubiquitinating Enzymes USP7 (also known as Herpes virus associated USP (HAUSP)) belongs to USP family of DUBs that has a critical role in cell proliferation and differentiation through regulating the activity of phosphatase and tensin homolog and localization of FOXO [60]. USP7 participates in p53 regulation and catalyzes deubiquitination of both p53 and Mdm2 [61, 62]. Therefore, USP7 can act as a tumor suppressor or an oncogene depending on whether it predominantly deubiquitinates p53 or Mdm2, respectively [63]. USP7 also deubiquitinates a tumor suppressor PTEN leading to cytoplasmic accumulation of this protein [60]. USP7 has been observed to be overexpressed in prostate cancer and associated with tumor aggressiveness [60], which makes this DUB is an important target for pharmaceutical intervention [59]. A high-throughput screen identified a small molecule USP7 inhibitor; HBX 41108 that can activate p53 resulting in p53-dependent apoptosis [64]. A fluorescencebased multiple assay has recently characterized another inhibitor (P022077) for USP7 [65]. Table 2. 5 Another member of USP family of DUBs is USP8 (UBPY), which functions in receptor endocytosis and trafficking processes and interact with several proteins including the epidermal growth factor receptor (EGFR) [66]. The depletion of USP8 leads to accumulation of ubiquitinated EGFR in endosomes [67]. Although an inhibitor of USP8; HBX 90397 is identified, the resulting effects are questionable since knockout of USP8 was observed to cause a liver failure in adult mice [67, 68]. USP2a deubiquitinates and stabilizes FAS, a metabolic oncogene overexpressed in many cancers [69]. The depletion of USP2a was observed to destabilize FAS and leads to apoptosis [69]. USP2a deubiquitinates Mdm2 as well, leading to stabilization of Mdm2 levels and induction of p53 degradation [70]. USP2a deubiquitinates also Mdmx, a Mdm2-like p53 repressor [71]. Interestingly, USP2a has been observed to have dual functions in disease. While overexpression of USP protects prostate cancer cells from apoptosis by reducing p53 stability, on the contrary, another study has illustrated that it is downregulated in breast carcinomas suggesting that USP2a may have an antitumor property [72]. USP28 is another cancer-associated DUB; it is overexpressed in breast and colon carcinomas, and is essential regulating the stability of c-Myc, a central modulator of cell growth, proliferation and apoptosis [73]. Since c-Myc is dysregulated in many human cancers and cancer cells require c-Myc activity for growth [74], inhibiting USP28 may be useful in targeting such malignancies by accelerating degradation of c-Myc [59]. USP14 is a DUB that blocks the degradation of ubiquitinated substrates. Loss of USP14 is related to neurodegenerative diseases resulting in an ataxic neurological phenotype in mice [75]. A smallmolecule inhibitor of USP14, called IU1, has been identified [76]. IU1 did not inhibit eight other tested deubiquitinating enzymes making it a relatively specific inhibitor of USP14 [76]. Treating the DUBs and Disease DUB Substrate Associated disease Alterations in disease Inhibitor A20 RIP, RIP2, TRAF6, NEMO and MALT1 [93-98] Several lymphomas including Hodgkin’s lymphoma and B cell lymphoma [204] Promoter methylation, mutations [204] - CYLD Bcl-3, TRAF2, TRAF6, TRAF7, RIP1, TAK1 [86-89] Kidney, colon, B-cell malignancies, multiple myeloma, uterine cervix carcinoma and melanoma [84] Mutations, decreased expression in cancer [84] - USP2a MDM2, FAS and MDMX [6971] Prostate cancer [205] Overexpression [205] - USP28 c-Myc [73], 53BP1 [206] Breast and colon cancers [73] Overexpression, somatic mutations in cancer [73] - USP7 p53, MDM2, PTEN, FOXO4 [60-62, 207, 208] Prostate, colon, non-small cell lung cancer [60, 82] Overexpression [60], USP7 inhibitor, HBX41108 activates p53 [64] and P022077 [65] USP8 Several receptor tyrosine kinases including MET, EGFR [66] Multiple abnormalities in RTK-dependent pathways [209] The depletion of USP8 leads to accumulation of ubiquitinated EGFR in endosomes [67] USP8 inhibitor, HBX 90397[67] USP9x Several proteins including catenin, Smad4 and MCL1 [7779] Colon, breast, small cell lung carcinoma, lymphoma [79, 210] Overexpression [79, 210] - USP14 Tau, ataxin-3 [76] Neurodegenerative diseases, ataxia [75] Loss of USP14 [75] USP14 inhibitor, IU1 [76] DUBs together with their substrates, associated disease, alterations and available inhibitors are listed. Downregulation [82] 6 Current Pharmaceutical Design, 2013, Vol. 19, No. 00 cells with IU1 increases the degradation of several substrates including Tau and ataxin-3, both of which have been implicated in neurodegenerative diseases [76]. Such inhibitors targeting USP14 could, therefore, potentially be used to reduce or eliminate misfolded and aggregated proteins that accumulate in neurodegenerative diseases and improve the prognosis in such disorders [25]. USP9x is another DUB from USP family that deubiquitinates several proteins related to cancer disease such as -catenin, Smad4 and MCL1 [77-79]. -catenin is known to promote colon cancer cell growth and it is stabilized by USP9x [77]. By an siRNA-based screen, USP9x has been found to be important for TGF signaling [78]. An essential component of TGF signaling pathway, Smad4, becomes inactive by undergoing monoubiquitination. USP9x was observed to deubiquitinate Smad4, hence, it positively regulates TGF signaling pathway, which is related to tumor initiation and progression [78]. USP9x also deubiquitinates MCL1, that is an antiapoptotic BCL-2 homolog leading to blocking of MCL1 degradation and stabilizing MCL1 levels, thus leading to cell survival [79]. However, having a dual function, it can also promote apoptosis by stabilizing apoptosis signal-regulating kinase 1 [80]. Other DUBs belonging to USP family that are linked to disease are: USP1 related to Fanconi anaemia DNA repair pathway [81]; USP6 to aneurismal bone cysts [58]; and USP7 to non-small cell lung adenocarcinoma [82]. UCH Family of Deubiquitinating Enzymes CYLD is a DUB belonging to UCH family and is mutated in Familial Cylindromatosis disease, in which numerous skin tumors develop [83]. Mutation or decreased expression levels of CYLD have been observed in kidney cancer, colon cancer, B-cell malignancies, multiple myeloma, uterine cervix carcinoma and melanoma [84]. CYLD deubiquitinates the Bcl-3 protein, which is a transcriptional co-activator of NF-B, blocks its nuclear translocation and hence inhibits NF-B activation [85]. CYLD also deubiquitinates TRAF2, TRAF6, TRAF7 (TNF Receptor Associated Factors), RIP1, TAK1 leading to inactivation of NF-B signaling [8689]. CYLD is also associated with Wnt signaling pathway that is essential for embryonic development and is frequently activated in cancer [90]. It negatively regulates Wnt signaling through deubiquitinating Dishevelled protein and activating -catenin [91]. Having a tumor suppressor role, increasing the activity of CYLD that is lost during tumor progression may be an efficient treatment in cancer. OTU Family of Deubiquitinating Enzymes A20 contains an OTU domain at the amino-terminus that possesses DUB activity. Additionally, it also functions as an E3 ligase through its carboxyl-terminal consisting of seven zinc fingers. A20 can act as tumor suppressor by downregulating NF-B signalling [92]. It removes K63-linked ubiquitin chains from RIP, RIP2, TRAF6, NEMO and MALT1 [93-98]. With its E3 ligase activity, it conjugates K48-linked polyubiquitin chains to RIP1 and TRAF2 targeting them for degradation [99]. Inactivation of A20 protein is associated with several chronic inflammatory and autoimmune diseases. JAMM Family of Deubiquitinating Enzymes The JAMM domain proteins are zinc metalloisopeptidases [58]. Members of this class include AMSH, BRCC36, CSN5 and POH1. The activity of AMSH regulates trafficking of G protein-coupled receptors and receptor tyrosine kinases [100]. BRCC36 has a role in the DNA damage response. CSN5 mediates NEDD8 attachment to cullins, regulating the cullin-RING ligase activity and stability . Finally, POH1 functions as a part of 19S cap of the 26S proteasome to trim polyubiquitin chains from the substrates. Though no drugs are yet available, these DUBs are potential drug targets. Kar et al. Targeting Proteasome The proteasome is a large, 26S, multicatalytic protease in which proteolysis of short-lived or regulatory proteins from the cytoplasm, the nucleus or the endoplasmic reticulum is carried out predominantly [101]. The 26S proteasome (proteasome holoenzyme) consists of two major subcomplexes: the 20S core particle (CP) that contains the protease subunits and the 19S regulatory particle (RP) that regulates the function of 20S CP [101]. The 19S regulatory particle contains two mutisubunit structures, a lid and a base. It has multiple roles in regulating proteasomal activity: selecting substrates, preparing them for degradation and translocating them into the CP. The proteolytic activity of the proteasome occurs in the 20S CP that contains two inner -rings that have the proteolytic active sites [101]. Active sites of 20S proteasome are the caspase-like (1), trypsin-like (2) and chymotrypsin-like (5) that use an amino terminal threonine as the catalytic aminoacid residue. There are one or two regulatory particles attached to the surface of the outer rings of the 20S CP to form 26S proteaome. After degradation of the substrate, short peptides derived from the substrate and reusable ubiquitin are released. Alterations in the proteasome are associated with many human diseases including neurodegenerative disorders, cardiac dysfunction, cataract formation, cachexia and rheumatoid diseases [102]. Several antiapoptotic and proliferative signaling pathways such as pro-oncogenic NF- pathway require proteasomal activity [4]. NF pathway is activated in many tumor types [103]. The inhibitors of NF-s (IBs) keep NF- factors in an inactive state. Upon phoshorylation and polyubiquitination of IBs and subsequent degradation by the proteaosome, NF- factors become activated [103]. The first clinically validated drug to target UPS is bortezomib, which reversibly inhibits the active sites in the 20S proteasome. Bortezomib-mediated proteasome inhibition leads to downregulation of NF- signaling through blocking IB degradation [104] and to endoplasmic reticulum stress, which in turn promotes cell death [105]. The effects of NF- inhibition are the upregulation of the cyclin-dependent kinase inhibitors p21 and p27 and downregulation of pro-inflammatory response genes, consequently leading to increased apoptosis in tumor cells [106]. Bortezomib is currently used as an anticancer drug for the treatment of multiple myeloma, relapsed mantle cell lymphoma. It is also in many other clinical trials including Phase III clinical trials for follicular nonHodgkin’s lymphoma and Phase II trials for diffuse large B cell lymphoma [25]. Owing to success of Bortezomib, several new generation of proteasome inhibitors such as Carfilzomib (PR-171), MLN9708, CEP18770, NPI-0052, ONX0912, PR957 and IPSI-001 are currently in development [107-114]. These proteasome inhibitors target different active sites of the 20S proteasome and differ in binding kinetics. For example, while Bortezomib inhibits the caspase-like (1) and chymotrypsin-like (5) activities of the proteasome in a reversible manner, NPI-0052 irreversibly blocks the trypsin-like (2) and chymotrypsin-like (5) sites. NPI-0052 is in Phase I stage and when combined with Bortezomib in low doses has been observed to show synergistic effects on cancer cells in vitro [115]. Another proteasome inhibitor, Carfilzomib, which has entered Phase III trials targets mainly the 5 site of the proteasome in an irreversible manner and has been developed for the treatment of relapsed multiple myeloma [107]. Other proteasome inhibitors are detailed in Table 3. Despite the success of Bortezomib and efforts for developing other inhibitors, targeting proteasome is somewhat problematic since this final common step of the degradative pathways are used by hundreds of different processes in the cell. Therefore, inhibition of the proteasome is likely to interrupt several diverse pathways. Emerging Role of the Ubiquitin-proteasome System Table 3. Current Pharmaceutical Design, 2013, Vol. 19, No. 00 7 Proteasome Inhibitors, Development Stage, Binding Kinetics and Targeted Disease Inhibitor Development Stage Disease Bortezomib Approved Multiple myeloma and mantle cell lymphoma Carfilzomib Phase III Multiple myeloma and other cancers Irreversible CEP18770 Phase I Multiple myeloma and other cancers Slowly reversible IPSI-001 Preclinical MLN9708 Phase I Multiple myeloma and other cancers Reversible NPI-0052 Phase I Multiple myeloma and leukemia Irreversible ONX0912 Phase I Multiple myeloma and other cancers Irreversible PR957 Preclinical Autoimmune disorders Irreversible Multiple myeloma and other hematologic malignancies Developing proteasome inhibitors with distinct substrate selectivity and lower toxicity would reduce such inconveniences in treating associated diseases. 3. IMPORTANCE OF STRUCTURES IN TARGETING UPS INTERACTIONS Protein-protein interactions have a central role in regulating many biological processes, cellular and signaling pathways. Alterations in protein-protein interactions may lead to several diseases such as cancer and neurological disorders. Therefore, proteinprotein interactions are recurrently considered as drug targets in disease states [116-122]. Drugs targeting protein-protein interactions, ultimately head protein interfaces where two proteins come into contact. Understanding the details and principles of protein interfaces is, therefore, immensely essential to develop efficient strategies in drug design. At protein interfaces there are some critical residues that account for the majority of the binding energy called hot spots [123]. A hot spot is defined as a residue that, when mutated to alanine, leads to a dramatic decrease in binding free energy (Gbinding > 2 kcal/mol) [123, 124]. Since these residues are more critical than others to the stability of the complex, targeting the hot spots may lead to improved inhibition of protein-protein Binding Kinetics Slowly reversible Not reported interactions and to designation of novel compounds. A potent inhibitor mimicking such key residue interactions can achieve favorable interaction energy; thus, can compete with the original partner for binding [121]. One such competitive inhibitor in treating immune disorders is SP-4206 that binds to hot spot residues of cytokine IL-2 with high affinity and block the formation of the natural complex of IL-2 and its receptor IL-2R [125]. The most widely known example of competitive inhibitors of the UPS is nutlins that prevent the Mdm2-p53 binding, thus enhance the tumor suppressor activity of p53. Alanine-scanning of the Mdm2-p53 binding region illustrated that there are three hot spot residues, Phe19, Trp23 and Leu26, on p53 critical for binding to Mdm2 [126]. Nutlin family of compounds such as Nutlin-2 and Nutlin-3 mimics p53 and binds to Mdm2 even with a greater affinity than p53 [127]. Mdm2-p53 and Mdm2-Nutlin-2 binding are visualized in (Fig. 2). In the UPS, there are five major steps: activation, conjugation, ubiquitin-conjugate recognition, ubiquitin removal, and substrate degradation by the proteasome [1]. Since the greatest amount of specificity is the conjugation step that is mainly mediated by E3s, the interactions of E3s represent important drug targets. The E3 and E2 proteins function in a concerted manner; however, the principles determining E2-E3 selectivity are still not entirely understood. In Fig. (2). Nutlin-2 targeting Mdm2-p53 interaction interface. (A) Mdm2 is an E3 ligase mediating ubiquitination and subsequent degradation of p53. Alanine-scanning of the Mdm2-p53 binding region illustrated that there are three hot spot residues, Phe19, Trp23 and Leu26, on p53 critical for binding to Mdm2 [126]. (B) Nutlin-2 prevents the interaction between Mdm2 and p53 by mimicking the conformation of Phe19, Trp23 and Leu26 of p53 to block Mdm2 p53-binding region [127]. 8 Current Pharmaceutical Design, 2013, Vol. 19, No. 00 addition, in many cases it is not known which E3s interact with which E2s and how they interact with each other. Identifying all E2 partners of a specific E3 is essential for inferring the principles guiding E2 selection by an E3. In one experimental approach, all E2 partners of Brca1 ubiquitin ligase were identified by two-hybrid experiments [128, 129]. Brca1 was found to interact with multiple different E2s and to possess E2-specific ubiquitin-transfer properties. Gardner et al. identified the interactions of HRD1 (HMG-CoA reductase degradation) ubiquitin ligase by in vivo cross-linking methods [130]. Ballar et al. illustrated how HRD1 can further act as an E3 for another E3, gp78 involved in ER-associated degradation [131]. Two large-scale studies have addressed this E2-E3 identification problem. In a global yeast-two hybrid screen, over 300 high quality interactions are uncovered [132]. They generated mutants of UBE2N to mimic the interaction pattern of another E2, UBE2D2, which resulted in new E3 interactions and hence illustrated how the E3 interactions of the E2 specific for K63 linkages UBE2N can be altered towards the K48-specific UBE2D2 [132]. Markson et al. combined yeast two-hybrid screens with homology modeling methods yielding a map of human E2-E3 RING interactions [133]. Their data includes 535 experimentally defined novel E2-RING E3 interactions and more than 1300 E2-RING E3 pairs with favorable predicted free energy values. They extended this network by including known human E2-RING E3 pairs, hence, assembled a network of 2644 proteins and 5087 interactions [133]. Including structural details of the interactions can assist in understanding principles beyond E3 selectivity. The first structural clues of E2-E3 interaction specificity were discovered from the crystal structure of the E3 ligase c-Cbl RING and the E2, UBE2L3 [134] (Fig. 3A). The -helix and zinc-chelating loops of the RING domain are in contact with loop L1 and L2 of UBE2L3. In particular, F63 residue in loop L1, P97 and A98 in loop L2 of UBE2L3 mediate its binding to c-Cbl. The c-Cbl linker region interacts with -helix 1 of UBE2L3 [134]. Currently, there are 9 E2-E3 complex structures deposited in PDB; UBE2D2/CNOT4 complex (pdb code: 1ur6:AB), Baculoviral IAP repeat-containing protein 3/ UBE2D2 (pdb code: 3eb6:AB), Signal transduction protein C-cbl/UBE2L3 Kar et al. complex (pdb code: 1fbv:AC), HECT(NEDD4L)/ UBE2D2 complex (pdb codes: 3jw0:AC and 3jvz:AC), TNF receptor-associated factor 6 (TRAF6)/UBE2N complex (pdb code: 3hct:AB), STIP1 homology and U-Box containing protein 1 (CHIP)/UBE2D1 complex (pdb code: 2oxq:AC), Ubiquitin-protein ligase Ube3a (E6AP)/ UBE2L3 complex (pdb code: 1c4z:AD) and U-box domain of human E4B ubiquitin ligase/UBE2D3 complex (pdb code: 3l1z:AB). Comparison of UBE2L3 binding to RING type c-Cbl and HECT type E6-AP revealed that there are common structural elements of this E2 recognized by both E3s: most extensive contacts are provided by F63, P97 and A98 residues of UBE2L3 binding to both RING type c-Cbl and HECT type E6-AP [134]. Comparison of all known E2-E3 structures through identifying the structurally conserved contacts implied that loop L1 second position residue contacts are only employed when interacting with HECT E3s [135]. Combining such structural details with protein interactions networks of UPS, obviously, improves drug design concept as outlined in the following section. Structural Interaction Network of UPS Proteins Network-based approaches have gained importance in drug discovery with the realization that diseases are complex as they are often consequences of multiple molecular abnormalities rather than being the result of a single defect or phenomena [136]. To go a step further in the mechanistic understanding one has to combine interaction networks information with molecular details of protein interfaces in the effort of identifying all target proteins that are influenced by a drug, either positively or negatively. The number of distinct binding motifs is limited in nature [137] implying that structurally different proteins can share similar interface architectures [138]. Following this notion, an algorithm called Prism has been developed that predicts potential protein associations based on known protein interface motifs [139, 140]. Exploiting this algorithm and the available structural proteome, recently, we have constructed a human E2-E3 structural interaction network [135]. Considering the all human genome, protein structures for 24 Fig. (3). Interactions of c-Cbl E3 ubiquitin ligase with two E2 proteins, UBE2L3 and UBE2D1. (A) c-Cbl interacts with UBE2L3 (pdb code: 1fbv:AC). helix 1, loop L1 and loop L2 regions of UBE2L3 are displayed in yellow, green and orange color, respectively. Two loop regions, L1 and L2 of UBE2L3 are in contact with the -helix and zinc-chelating loops of the RING domain. The central F63 residue in loop L1, P97 and A98 in loop L2 of UBE2L3 mediate its binding to c-Cbl. The c-Cbl linker region interacts with -helix 1 of UBE2L3 [134]. (B) Modeled c-Cbl-UBE2D1 complex. Although the interaction was reported earlier [157], binding details and the structure of the complex was not available. c-Cbl residues labeled in red color was observed to be critical: C384, C404, R420 were mutated in patients with acute myelogenous leukemia [158]. Additionally, Ile383 and Trp408 of c-Cbl were shown to have a critical role in E2 binding [134]. These residues correspond to computational hotspots predicted by Hotpoint server indicating the usefulness of structural modeling of E2-E3 associations. Emerging Role of the Ubiquitin-proteasome System E2s are available (out of ~40) while the structural coverage is relatively low for E3 proteins; 32 of them (out of ~500) have resolved structures in PDB. Among these, we predict 107 interactions between 22 E2s and 16 E3s. In our network, the number of all possible E2-E3 pairs is 352 (i.e. 22x16) and 51 of these were already reported in earlier studies. We recover 76% (39/51) of the known pairs through 107 predicted interactions verifying 36% (39/107) of the network. On the basis of known E2-E3 interaction data, the accuracy of the predictions is 76% indicating that the predicted E2E3 interactions are in agreement with validated functional E2-E3 pairs. Our method depends on the coverage of known interface architectures and there are only 9 E2-E3 known complex structures in the PDB (as aforementioned above) which are used as a basis for modeling. Because the size and coverage of the PDB increases exponentially, we expect the prediction efficiency of our method to increase. We observed that E3 proteins could interact with multiple E2s and likewise E2 proteins could interact with multiple E3s, as expected. To reveal which residues are conserved among E2s interacting with a common E3 and which residues differ, hence, to provide clues to specificity, we analyzed the E2-E3 interfaces and predicted hotspot residues at interfaces using the Hotpoint server [141]. The Hotpoint server determines computational hot spot residues based on conservation, solvent accessibility and statistical pairwise residue potentials of the interface residues. The predicted hot spots are observed to match with the experimental hot spots with an accuracy of 70% [141]. Considering E2-E3 interface regions, some residues are structurally conserved among E2 proteins and appear to be essential for all E2-E3 interactions, whereas others, particularly in loop L1, appear to play important roles in E3 selectivity. The E2 proteins which have been characterized so far are known to recognize E3s through the L1 and L2 loops and the N-terminal -helix 1 on the E2 surface [142]. In particular, loop L1 has been shown to be critical in E3 binding. In our network, E2 interface residues also mostly correspond to the -helix 1, loop L1 and L2 regions. The residue in the sixth position in loop L1 is widely utilized as an interface hot spot and appears indispensible for E2 interactions. Other loop L1 residues also confer specificity on the E2-E3 interactions: HECT E3s are in contact with the residue in the second position in loop L1 of E2s; but this is not the case for the RING finger type E3s [135]. Knowledge of such structural details and all interacting partners of E3s at a proteome-level are crucial in drug design. This effort will aid in explaining: i) which E3s interact with which E2s and which substrates; ii) the molecular determinants of these interactions; iii) what the corresponding functions and outcomes are. In the drug design field, to date, researchers have focused primarily on blocking E3-substrate interfaces (as aforementioned above). However, targeting and disrupting E2-E3 interactions may be relatively easy since these interactions are usually relatively weak [142]. Such inhibitors can block the interaction by binding to the E2, the E3 or the E2-E3 interface. Structural examples showing the binding of the inhibitors are provided as case studies in the next section. Flexibility and Molecular Dynamics of UPS Proteins It is more and more recognized that increment in protein flexibility is paralleled by an increment in binding promiscuity of the flexible region. This is particularly true for those region that undergo post-translational modifications such as phosphorylation [143, 144] or ubiquitination [145, 146] and for their partners [147, 148]. These flexible changes are mostly studied by NMR and Molecular Dynamics (MD) [149]. Studies using NMR residual dipolar couplings has shown neatly that free ubiquitin samples conformations globally similar to those in the bound state [150, 151]. Lange and colleagues compared a number of X-ray structures of ubiquitin, bound to different partners, with NMR structures of the ubiquitin protein free in solution. In this pioneering study the authors were Current Pharmaceutical Design, 2013, Vol. 19, No. 00 9 able to demonstrate, by analyzing the dynamic behavior of ubiquitin from the the picosecond to microsecond time scale, that for each bound ubiquitin structure there is a member of the unbound sampled ensemble that is structurally similar to it [151]. The work strongly supported an underlying conformational selection mechanism for these multibinding promiscuous proteins. Lately, by Molecular Dynamics investigations of the same data by Wlodarski and Zagrovic suggested that during the binding event the conformation structurally most similar to the bound structure is the dominant one via the conformational selection model. After this selection, reoptimization of the binding interface occurs via a ‘residual induced fit’ mechanism, therefore calling in cause both mechanisms of binding for these enzymes [152]. Long and Brüschweiler performed microsecond time scale all-atom MD simulations of ubiquitin and its binding partners using the latest generation of molecular mechanics force field [153]. In this study, the effect of ligand molecules on the flexibility and plasticity of ubiquitin binding properties is analyzed. The study focuses on the ubiquitin interacting motif (UIM), a short alpha-helical conserved motif for ubiquitin recognition. The energy landscape and dynamics of ubiquitin in response to the approach of the UIM ligand is analyzed in details. By the use of Boltzmann sampling of molecular dynamics snapshots, the authors show a statistical reweighing of the populations of ubiquitin conformers in the presence of its ligand molecule at intermediate distance range (1–2 nm) and are able to examine the population redistribution mechanisms. Nussinov and collaborators focused on cullin-RING E3 ubiquitin ligases and their flexible role in binding to many partners [145, 146]. They performed structural analysis and MD simulations starting from Cul1, Cul4A, and Cul5 crystal structures. The MD work clearly shows that at least three cullins do not act as merely rigid scaffolds but are flexible with conserved hinges in the Nterminal domain. In another study, dynamical properties of the E2 enzymes of family 3R are characterized, showing a structural role of Loop 7 and Loop 8 in stabilizing closed conformations [154]. Taken together, all these evidences highlight the importance of flexibility in the recognition and functional activity of these E3 ligase enzymes and pave the way for more studies in which Molecular Dynamics techniques are used for the conformational characterization and identification of flexible regions to be targeted in drug design. 4. CASE STUDIES Here we present two case studies of UPS proteins relevant to drug design. The first example illustrates how structural modeling of E2-E3 associations may be beneficial to drug discovery. In the second example, an interesting approach of designing a smallmolecule antagonist to target an E3 protein in cancer is explained. Modeling E2-E3 Complexes: UBE2D1-c-Cbl Ligase Example c-Cbl is an E3 ubiquitin ligase that attenuates signaling through poly- or monoubiquitination of several activated receptor tyrosine kinases (such as Flt-3, c-kit, and M-CSF) and other tyrosine kinases of the Scr family [155]. The RING domain has a central role in cCbl function because its deletion or disruption abolishes the function of c-Cbl [156]. From its RING domain, c-Cbl binds to an E2 and mediates ubiquitin transfer from E2 to the target substrates. Structure of c-Cbl interacting with one of its E2 partners, UBE2L3, is deposited in Protein Data Bank (PDB) (pdb code: 1fbv) [134]. Two loop regions, L1 and L2 of UBE2L3 are in contact with the helix and zinc-chelating loops of the RING domain. The central F63 residue in loop L1, P97 and A98 in loop L2 of UBE2L3 mediate its binding to c-Cbl. The c-Cbl linker region interacts with helix 1 of UBE2L3 [134]. c-Cbl ligase-UBE2L3 complex showing the critical binding regions (-helix 1, loop L1 and L2) of UBE2L3 is visualized in (Fig. 3A). 10 Current Pharmaceutical Design, 2013, Vol. 19, No. 00 c-Cbl is also reported to interact with another E2, UBE2D1, and ubiquitinate epidermal growth factor receptor [157], although the structure of the complex is not available as yet. Using Prism algorithm, we modeled c-Cbl-UBE2D1 complex based on a known interface of another E2-E3 complex; UBE2D2-CNOT4 (pdb code: 1ur6:AB) [135]. We observe that UBE2D1 (pdb code: 2c4p:A) binds to c-Cbl (pdb code: 1fbv:A) mainly through its -helix 1, loop L1 and L2 regions. Earlier work suggested that mutations in the cCbl RING domain are associated with acute myelogenous leukemia and myeloproliferative neoplasms and the impairing the degradation of tyrosine kinases is an important mechanism in cancer [158]. In particular, on the RING domain of c-Cbl, the substitution of cysteine or arginine residues at position 384 (C384Y), 404 (C404S), and 420 (R420Q) are observed in patients with acute myelogenous leukemia [158]. Additionally, Ile383 and Trp408 of c-Cbl were shown to have a critical role in E2 binding [134] and the mutation of Trp408 to alanine reduces c-Cbl’s affinity for the E2 and eliminates its ubiquitin-ligase activity in vitro [159]. Interface analysis of our c-Cbl-UBE2D1 model indicates that these critical residues; I383, C384, C404 and W408, correspond to computational hotspots predicted by Hotpoint server [141], implying their importance in binding and function of c-Cbl. Such structural modeling of E2-E3 associations could help in understanding E2-E3 selectivity and discovering drug candidates targeting E3s. (Fig. 3B) illustrates the modeled c-Cbl-UBE2D1 complex and the critical residues in binding. A Potent Small Molecule Antagonist of IAP Proteins: GDC0152 Inhibitors of apoptosis (IAP) family of proteins function in the regulation of signal transduction pathways associated with malignancy [160, 161] and are frequently overexpressed in cancer cells [162, 163]. Cellular IAP (cIAP) proteins have been observed to regulate TNF-mediated NF-B activation via its RING domain and ubiquitinate receptor interacting protein RIP-1 and NF-B inducing kinase, NIK [164]. Viability of cancer cells depend on the aberrations in the apoptosis signaling pathways. Therefore, drugs that can restore apoptosis in tumor cells might be efficient in treating cancer [165]. In cancer, one approach to target the IAP proteins is to design of small Kar et al. molecules that mimic the binding of IAP antagonist, second mitochondria-derived activator of caspases/direct IAP-binding protein with low pI (SMAC/DIABLO) [166, 167], to baculoviral IAP repeat (BIR) domains [168]. The IAP BIR domains are necessary for the antiapoptotic function of the IAP proteins [165]. One such antagonist is GDC-0152, which has entered Phase I clinical trials, can bind to BIR3 domain of cIAP1 [165]. Binding of GDC-0152 to cIAP1 promotes degradation of cIAP1, induces activation of caspase-3/7, and leads to decreased viability of breast cancer cells without affecting normal mammary epithelial cells [165]. (Fig. 4A) illustrates N-terminal peptide from SMAC/DIABLO binding to BIR3 domain of cIAP1 (pdb code: 3d9u). GDC-0152 antagonist (pdb code: 3uw4), mimicking SMAC/DIABLO binding to cIAP1, is visualized in (Fig. 4B). With the complete knowledge of structural and binding details of UPS proteins, it will become undoubtedly easier to design such small-molecule compounds. 5. CONCLUSIONS Since the approval of the proteasome inhibitor Bortezomib in 2003, UPS components have emerged as crucial drug targets in treatment of cancer, neurodegenenerative diseases, immunological disorders and microbial infection. Small-molecule inhibitors of ubiquitin-conjugating and –deconjugating enzymes are continuously being developed and evaluated for their potential use as therapeutics. The major difficulty is, however, to design targetspecific inhibitors since UPS has diverse roles affecting hundreds of different processes in the cell. Although attracted little attention so far, developing selective compounds targeting E2-E3 interactions would be a crucial potential therapeutic intervention: inhibitors can be designed to bind to E3 ligases proximal to E2 binding region, thus blocking E2-E3 interface or disrupt E2-E3 interactions allosterically by inducing long-range conformational changes. At this point, knowledge of structural details of the interactions in UPS, especially at a proteome-scale, appears to be essential. In addition to targeting UPS with a single small-molecule compound, combinatorial drug therapy in UPS can be also efficient since drug combinations currently provide a route to overcome the complexity of human cancers [169]. Novel approaches including large-scale genomics, systems and structural biology will set the basis for future potential therapeutic interventions in UPS- Fig. (4). A potent small molecule antagonist targeting an E3, cIAP1 in cancer treatment. (A) cIAP1 interacting with N-terminal peptide from SMAC/DIABLO (pdb code: 3d9u). (B) GDC-0152 antagonist mimicks SMAC/DIABLO binding to cIAP1 (pdb code: 3uw4). Binding of GDC-0152 to cIAP1 promotes degradation of cIAP1, induces activation of caspase-3/7, and leads to decreased viability of breast cancer cells without affecting normal mammary epithelial cells [165]. Emerging Role of the Ubiquitin-proteasome System associated diseases and guide the identification of best possible drug combinations targeting UPS. CONFLICT OF INTEREST The authors confirm that this article content has no conflicts of interest. ACKNOWLEDGEMENTS Declared none. Current Pharmaceutical Design, 2013, Vol. 19, No. 00 [25] [26] [27] [28] [29] REFERENCES [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] [13] [14] [15] [16] [17] [18] [19] [20] [21] [22] [23] [24] Nalepa G, Rolfe M, Harper JW. Drug discovery in the ubiquitinproteasome system. Nat Rev Drug Discov 2006; 5: 596-613. Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem 2001; 70: 503-33. Bennett EJ, Harper JW. DNA damage: ubiquitin marks the spot. Nat Struct Mol Biol 2008; 15: 20-2. Hoeller D, Dikic I. Targeting the ubiquitin system in cancer therapy. Nature 2009; 458: 438-44. Herrmann J, Ciechanover A, Lerman LO, Lerman A. The ubiquitin-proteasome system in cardiovascular diseases-a hypothesis extended. Cardiovasc Res 2004; 61: 11-21. Petroski MD. The ubiquitin system, disease, and drug discovery. BMC Biochem 2008; 9 Suppl 1: S7. Lehman NL. The ubiquitin proteasome system in neuropathology. Acta Neuropathol 2009; 118: 329-47. Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem 1998; 67: 425-79. Yang Y, Kitagaki J, Dai RM, et al. Inhibitors of ubiquitinactivating enzyme (E1), a new class of potential cancer therapeutics. Cancer Res 2007; 67: 9472-81. Xu GW, Ali M, Wood TE, et al. The ubiquitin-activating enzyme E1 as a therapeutic target for the treatment of leukemia and multiple myeloma. Blood 2010; 115: 2251-9. Soucy TA, Smith PG, Milhollen MA, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 2009; 458: 732-6. Soucy TA, Smith PG, Rolfe M. Targeting NEDD8-activated cullinRING ligases for the treatment of cancer. Clin Cancer Res 2009; 15: 3912-6. van Ree JH, Jeganathan KB, Malureanu L, van Deursen JM. Overexpression of the E2 ubiquitin-conjugating enzyme UbcH10 causes chromosome missegregation and tumor formation. J Cell Biol 2010; 188: 83-100. Hao J, Xu A, Xie X, et al. Elevated expression of UBE2T in lung cancer tumors and cell lines. Tumour Biol 2008; 29: 195-203. Maeda H, Miyajima N, Kano S, et al. Ubiquitin-conjugating enzyme UBE2Q2 suppresses cell proliferation and is downregulated in recurrent head and neck cancer. Mol Cancer Res 2009; 7: 1553-62. Zhu S, Sachdeva M, Wu F, Lu Z, Mo YY. Ubc9 promotes breast cell invasion and metastasis in a sumoylation-independent manner. Oncogene 2010; 29: 1763-72. Duan X, Trent JO, Ye H. Targeting the SUMO E2 conjugating enzyme Ubc9 interaction for anti-cancer drug design. Anticancer Agents Med Chem 2009; 9: 51-4. Wong BR, Parlati F, Qu K, et al. Drug discovery in the ubiquitin regulatory pathway. Drug Discov Today 2003; 8: 746-54. Dominguez C, Bonvin AM, Winkler GS, et al. Structural model of the UbcH5B/CNOT4 complex revealed by combining NMR, mutagenesis, and docking approaches. Structure 2004; 12: 633-44. Weissman AM. Themes and variations on ubiquitylation. Nat Rev Mol Cell Biol 2001; 2: 169-78. Huang DT, Miller DW, Mathew R, et al. A unique E1-E2 interaction required for optimal conjugation of the ubiquitin-like protein NEDD8. Nat Struct Mol Biol 2004; 11: 927-35. Rotin D, Kumar S. Physiological functions of the HECT family of ubiquitin ligases. Nat Rev Mol Cell Biol 2009; 10: 398-409. Borden KL. RING domains: master builders of molecular scaffolds? J Mol Biol 2000; 295: 1103-12. Ohi MD, Vander Kooi CW, Rosenberg JA, Chazin WJ, Gould KL. Structural insights into the U-box, a domain associated with multiubiquitination. Nat Struct Biol 2003; 10: 250-5. [30] [31] [32] [33] [34] [35] [36] [37] [38] [39] [40] [41] [42] [43] [44] [45] [46] [47] [48] [49] [50] [51] [52] [53] 11 Cohen P, Tcherpakov M. Will the ubiquitin system furnish as many drug targets as protein kinases? Cell 2010; 143: 686-93. Quesnel B, Preudhomme C, Fournier J, Fenaux P, Peyrat JP. MDM2 gene amplification in human breast cancer. Eur J Cancer 1994; 30A: 982-4. Momand J, Jung D, Wilczynski S, Niland J. The MDM2 gene amplification database. Nucleic Acids Res 1998; 26: 3453-9. Vassilev LT, Vu BT, Graves B, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004; 303: 844-8. Issaeva N, Bozko P, Enge M, et al. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat Med 2004; 10: 1321-8. Bedford L, Lowe J, Dick LR, Mayer RJ, Brownell JE. Ubiquitinlike protein conjugation and the ubiquitin-proteasome system as drug targets. Nat Rev Drug Discov 2011; 10: 29-46. Merlet J, Burger J, Gomes JE, Pintard L. Regulation of cullinRING E3 ubiquitin-ligases by neddylation and dimerization. Cell Mol Life Sci 2009; 66: 1924-38. Chen Q, Xie W, Kuhn DJ, et al. Targeting the p27 E3 ligase SCF(Skp2) results in p27- and Skp2-mediated cell-cycle arrest and activation of autophagy. Blood 2008; 111: 4690-9. Nakajima H, Fujiwara H, Furuichi Y, Tanaka K, Shimbara N. A novel small-molecule inhibitor of NF-kappaB signaling. Biochem Biophys Res Commun 2008; 368: 1007-13. Orlicky S, Tang X, Neduva V, et al. An allosteric inhibitor of substrate recognition by the SCF(Cdc4) ubiquitin ligase. Nat Biotechnol 2010; 28: 733-7. Wu H, Tschopp J, Lin SC. Smac mimetics and TNFalpha: a dangerous liaison? Cell 2007; 131: 655-8. Ford D, Easton DF, Stratton M, et al. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium. Am J Hum Genet 1998; 62: 676-89. Scully R. Role of BRCA gene dysfunction in breast and ovarian cancer predisposition. Breast Cancer Res 2000; 2: 324-30. Ohta T, Fukuda M. Ubiquitin and breast cancer. Oncogene 2004; 23: 2079-88. Dawson TM, Dawson VL. The role of parkin in familial and sporadic Parkinson's disease. Mov Disord 2010; 25 Suppl 1: S32-9. Bedford L, Hay D, Devoy A, et al. Depletion of 26S proteasomes in mouse brain neurons causes neurodegeneration and Lewy-like inclusions resembling human pale bodies. J Neurosci 2008; 28: 8189-98. Lonser RR, Glenn GM, Walther M, et al. von Hippel-Lindau disease. Lancet 2003; 361: 2059-67. Maxwell PH, Wiesener MS, Chang GW, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999; 399: 271-5. Kim WY, Kaelin WG. Role of VHL gene mutation in human cancer. J Clin Oncol 2004; 22: 4991-5004. Sufan RI, Moriyama EH, Mariampillai A, et al. Oxygenindependent degradation of HIF-alpha via bioengineered VHL tumour suppressor complex. EMBO Mol Med 2009; 1: 66-78. Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. The HPV16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 1993; 75: 495-505. zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer 2002; 2: 342-50. Matsuura T, Sutcliffe JS, Fang P, et al. De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat Genet 1997; 15: 74-7. Chen D, Kon N, Li M, et al. ARF-BP1/Mule is a critical mediator of the ARF tumor suppressor. Cell 2005; 121: 1071-83. Chen D, Brooks CL, Gu W. ARF-BP1 as a potential therapeutic target. Br J Cancer 2006; 94: 1555-8. Wang X, Trotman LC, Koppie T, et al. NEDD4-1 is a protooncogenic ubiquitin ligase for PTEN. Cell 2007; 128: 129-39. Fouladkou F, Landry T, Kawabe H, et al. The ubiquitin ligase Nedd4-1 is dispensable for the regulation of PTEN stability and localization. Proc Natl Acad Sci U S A 2008; 105: 8585-90. Baker SJ. PTEN enters the nuclear age. Cell 2007; 128: 25-8. Zhang Y, Chang C, Gehling DJ, Hemmati-Brivanlou A, Derynck R. Regulation of Smad degradation and activity by Smurf2, an E3 ubiquitin ligase. Proc Natl Acad Sci U S A 2001; 98: 974-9. 12 Current Pharmaceutical Design, 2013, Vol. 19, No. 00 [54] [55] [56] [57] [58] [59] [60] [61] [62] [63] [64] [65] [66] [67] [68] [69] [70] [71] [72] [73] [74] [75] [76] [77] Lin X, Liang M, Feng XH. Smurf2 is a ubiquitin E3 ligase mediating proteasome-dependent degradation of Smad2 in transforming growth factor-beta signaling. J Biol Chem 2000; 275: 36818-22. Fukuchi M, Fukai Y, Masuda N, et al. High-level expression of the Smad ubiquitin ligase Smurf2 correlates with poor prognosis in patients with esophageal squamous cell carcinoma. Cancer Res 2002; 62: 7162-5. Chen C, Zhou Z, Sheehan CE, et al. Overexpression of WWP1 is associated with the estrogen receptor and insulin-like growth factor receptor 1 in breast carcinoma. Int J Cancer 2009; 124: 2829-36. Laine A, Ronai Z. Regulation of p53 localization and transcription by the HECT domain E3 ligase WWP1. Oncogene 2007; 26: 147783. Singhal S, Taylor MC, Baker RT. Deubiquitylating enzymes and disease. BMC Biochem 2008; 9 Suppl 1: S3. Shi D, Grossman SR. Ubiquitin becomes ubiquitous in cancer: emerging roles of ubiquitin ligases and deubiquitinases in tumorigenesis and as therapeutic targets. Cancer Biol Ther 2010; 10: 737-47. Song MS, Salmena L, Carracedo A, et al. The deubiquitinylation and localization of PTEN are regulated by a HAUSP-PML network. Nature 2008; 455: 813-7. Li M, Brooks CL, Kon N, Gu W. A dynamic role of HAUSP in the p53-Mdm2 pathway. Mol Cell 2004; 13: 879-86. Li M, Chen D, Shiloh A, et al. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature 2002; 416: 648-53. Fraile JM, Quesada V, Rodriguez D, Freije JM, Lopez-Otin C. Deubiquitinases in cancer: new functions and therapeutic options. Oncogene 2012; 31: 2373-88. Colland F, Formstecher E, Jacq X, et al. Small-molecule inhibitor of USP7/HAUSP ubiquitin protease stabilizes and activates p53 in cells. Mol Cancer Ther 2009; 8: 2286-95. Tian X, Isamiddinova NS, Peroutka RJ, et al. Characterization of selective ubiquitin and ubiquitin-like protease inhibitors using a fluorescence-based multiplex assay format. Assay Drug Dev Technol 2011; 9: 165-73. Mizuno E, Kobayashi K, Yamamoto A, Kitamura N, Komada M. A deubiquitinating enzyme UBPY regulates the level of protein ubiquitination on endosomes. Traffic 2006; 7: 1017-31. Colombo M, Vallese S, Peretto I, et al. Synthesis and biological evaluation of 9-oxo-9H-indeno[1,2-b]pyrazine-2,3-dicarbonitrile analogues as potential inhibitors of deubiquitinating enzymes. ChemMedChem 2010; 5: 552-8. Niendorf S, Oksche A, Kisser A, et al. Essential role of ubiquitinspecific protease 8 for receptor tyrosine kinase stability and endocytic trafficking in vivo. Mol Cell Biol 2007; 27: 5029-39. Graner E, Tang D, Rossi S, et al. The isopeptidase USP2a regulates the stability of fatty acid synthase in prostate cancer. Cancer Cell 2004; 5: 253-61. Stevenson LF, Sparks A, Allende-Vega N, et al. The deubiquitinating enzyme USP2a regulates the p53 pathway by targeting Mdm2. EMBO J 2007; 26: 976-86. Allende-Vega N, Sparks A, Lane DP, Saville MK. MdmX is a substrate for the deubiquitinating enzyme USP2a. Oncogene 2010; 29: 432-41. Metzig M, Nickles D, Falschlehner C, et al. An RNAi screen identifies USP2 as a factor required for TNF-alpha-induced NFkappaB signaling. Int J Cancer 2011; 129: 607-18. Popov N, Wanzel M, Madiredjo M, et al. The ubiquitin-specific protease USP28 is required for MYC stability. Nat Cell Biol 2007; 9: 765-74. Albihn A, Johnsen JI, Henriksson MA. MYC in oncogenesis and as a target for cancer therapies. Adv Cancer Res 2010; 107: 163-224. Anderson C, Crimmins S, Wilson JA, et al. Loss of Usp14 results in reduced levels of ubiquitin in ataxia mice. J Neurochem 2005; 95: 724-31. Lee BH, Lee MJ, Park S, et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature 2010; 467: 179-84. Murray RZ, Jolly LA, Wood SA. The FAM deubiquitylating enzyme localizes to multiple points of protein trafficking in epithelia, where it associates with E-cadherin and beta-catenin. Mol Biol Cell 2004; 15: 1591-9. Kar et al. [78] [79] [80] [81] [82] [83] [84] [85] [86] [87] [88] [89] [90] [91] [92] [93] [94] [95] [96] [97] [98] [99] [100] [101] Dupont S, Mamidi A, Cordenonsi M, et al. FAM/USP9x, a deubiquitinating enzyme essential for TGFbeta signaling, controls Smad4 monoubiquitination. Cell 2009; 136: 123-35. Schwickart M, Huang X, Lill JR, et al. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature 2010; 463: 103-7. Nagai H, Noguchi T, Homma K, et al. Ubiquitin-like sequence in ASK1 plays critical roles in the recognition and stabilization by USP9X and oxidative stress-induced cell death. Mol Cell 2009; 36: 805-18. Cohn MA, Kowal P, Yang K, et al. A UAF1-containing multisubunit protein complex regulates the Fanconi anemia pathway. Mol Cell 2007; 28: 786-97. Masuya D, Huang C, Liu D, et al. The HAUSP gene plays an important role in non-small cell lung carcinogenesis through p53dependent pathways. J Pathol 2006; 208: 724-32. Bignell GR, Warren W, Seal S, et al. Identification of the familial cylindromatosis tumour-suppressor gene. Nat Genet 2000; 25: 1605. Sun SC. CYLD: a tumor suppressor deubiquitinase regulating NFkappaB activation and diverse biological processes. Cell Death Differ 2010; 17: 25-34. Massoumi R, Chmielarska K, Hennecke K, Pfeifer A, Fassler R. Cyld inhibits tumor cell proliferation by blocking Bcl-3-dependent NF-kappaB signaling. Cell 2006; 125: 665-77. Brummelkamp TR, Nijman SM, Dirac AM, Bernards R. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-kappaB. Nature 2003; 424: 797-801. Kovalenko A, Chable-Bessia C, Cantarella G, et al. The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature 2003; 424: 801-5. Reiley WW, Jin W, Lee AJ, et al. Deubiquitinating enzyme CYLD negatively regulates the ubiquitin-dependent kinase Tak1 and prevents abnormal T cell responses. J Exp Med 2007; 204: 147585. Yoshida H, Jono H, Kai H, Li JD. The tumor suppressor cylindromatosis (CYLD) acts as a negative regulator for toll-like receptor 2 signaling via negative cross-talk with TRAF6 AND TRAF7. J Biol Chem 2005; 280: 41111-21. Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer 2008; 8: 387-98. Tauriello DV, Haegebarth A, Kuper I, et al. Loss of the tumor suppressor CYLD enhances Wnt/beta-catenin signaling through K63-linked ubiquitination of Dvl. Mol Cell 2010; 37: 607-19. Wertz IE, O'Rourke KM, Zhou H, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature 2004; 430: 694-9. Hitotsumatsu O, Ahmad RC, Tavares R, et al. The ubiquitinediting enzyme A20 restricts nucleotide-binding oligomerization domain containing 2-triggered signals. Immunity 2008; 28: 381-90. Turer EE, Tavares RM, Mortier E, et al. Homeostatic MyD88dependent signals cause lethal inflamMation in the absence of A20. J Exp Med 2008; 205: 451-64. Duwel M, Welteke V, Oeckinghaus A, et al. A20 negatively regulates T cell receptor signaling to NF-kappaB by cleaving Malt1 ubiquitin chains. J Immunol 2009; 182: 7718-28. Boone DL, Turer EE, Lee EG, et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol 2004; 5: 1052-60. Mauro C, Pacifico F, Lavorgna A, et al. ABIN-1 binds to NEMO/IKKgamma and co-operates with A20 in inhibiting NFkappaB. J Biol Chem 2006; 281: 18482-8. Lee EG, Boone DL, Chai S, et al. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science 2000; 289: 2350-4. Song HY, Rothe M, Goeddel DV. The tumor necrosis factorinducible zinc finger protein A20 interacts with TRAF1/TRAF2 and inhibits NF-kappaB activation. Proc Natl Acad Sci U S A 1996; 93: 6721-5. Sierra MI, Wright MH, Nash PD. AMSH interacts with ESCRT-0 to regulate the stability and trafficking of CXCR4. J Biol Chem 2010; 285: 13990-4004. Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev 2002; 82: 373-428. Emerging Role of the Ubiquitin-proteasome System [102] [103] [104] [105] [106] [107] [108] [109] [110] [111] [112] [113] [114] [115] [116] [117] [118] [119] [120] [121] [122] [123] [124] [125] [126] Dahlmann B. Role of proteasomes in disease. BMC Biochem 2007; 8 Suppl 1: S3. Karin M. Nuclear factor-kappaB in cancer development and progression. Nature 2006; 441: 431-6. Adams J. The development of proteasome inhibitors as anticancer drugs. Cancer Cell 2004; 5: 417-21. Gu H, Chen X, Gao G, Dong H. Caspase-2 functions upstream of mitochondria in endoplasmic reticulum stress-induced apoptosis by bortezomib in human myeloma cells. Mol Cancer Ther 2008; 7: 2298-307. Ling YH, Liebes L, Jiang JD, et al. Mechanisms of proteasome inhibitor PS-341-induced G(2)-M-phase arrest and apoptosis in human non-small cell lung cancer cell lines. Clin Cancer Res 2003; 9: 1145-54. Demo SD, Kirk CJ, Aujay MA, et al. Antitumor activity of PR171, a novel irreversible inhibitor of the proteasome. Cancer Res 2007; 67: 6383-91. Zhou HJ, Aujay MA, Bennett MK, et al. Design and synthesis of an orally bioavailable and selective peptide epoxyketone proteasome inhibitor (PR-047). J Med Chem 2009; 52: 3028-38. Kupperman E, Lee EC, Cao Y, et al. Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. Cancer Res 2010; 70: 1970-80. Piva R, Ruggeri B, Williams M, et al. CEP-18770: A novel, orally active proteasome inhibitor with a tumor-selective pharmacologic profile competitive with bortezomib. Blood 2008; 111: 2765-75. Dorsey BD, Iqbal M, Chatterjee S, et al. Discovery of a potent, selective, and orally active proteasome inhibitor for the treatment of cancer. J Med Chem 2008; 51: 1068-72. Muchamuel T, Basler M, Aujay MA, et al. A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nat Med 2009; 15: 781-7. Kuhn DJ, Hunsucker SA, Chen Q, et al. Targeted inhibition of the immunoproteasome is a potent strategy against models of multiple myeloma that overcomes resistance to conventional drugs and nonspecific proteasome inhibitors. Blood 2009; 113: 4667-76. Feling RH, Buchanan GO, Mincer TJ, et al. Salinosporamide A: a highly cytotoxic proteasome inhibitor from a novel microbial source, a marine bacterium of the new genus salinospora. Angew Chem Int Ed Engl 2003; 42: 355-7. Chauhan D, Singh A, Brahmandam M, et al. Combination of proteasome inhibitors bortezomib and NPI-0052 trigger in vivo synergistic cytotoxicity in multiple myeloma. Blood 2008; 111: 1654-64. Arkin MR, Wells JA. Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nat Rev Drug Discov 2004; 3: 301-17. Chene P. Drugs targeting protein-protein interactions. ChemMedChem 2006; 1: 400-11. Fry DC. Protein-protein interactions as targets for small molecule drug discovery. Biopolymers 2006; 84: 535-52. Gonzalez-Ruiz D, Gohlke H. Targeting protein-protein interactions with small molecules: challenges and perspectives for computational binding epitope detection and ligand finding. Curr Med Chem 2006; 13: 2607-25. Kar G, Kuzu G, Keskin O, Gursoy A. Protein-protein interfaces integrated into interaction networks: implications on drug design. Curr Pharm Des 2012; Keskin O, Gursoy A, Ma B, Nussinov R. Towards drugs targeting multiple proteins in a systems biology approach. Curr Top Med Chem 2007; 7: 943-51. Zhao L, Chmielewski J. Inhibiting protein-protein interactions using designed molecules. Curr Opin Struct Biol 2005; 15: 31-4. Bogan AA, Thorn KS. Anatomy of hot spots in protein interfaces. J Mol Biol 1998; 280: 1-9. Thorn KS, Bogan AA. ASEdb: a database of alanine mutations and their effects on the free energy of binding in protein interactions. Bioinformatics 2001; 17: 284-5. Thanos CD, DeLano WL, Wells JA. Hot-spot mimicry of a cytokine receptor by a small molecule. Proc Natl Acad Sci U S A 2006; 103: 15422-7. Picksley SM, Vojtesek B, Sparks A, Lane DP. Immunochemical analysis of the interaction of p53 with MDM2;--fine mapping of the MDM2 binding site on p53 using synthetic peptides. Oncogene 1994; 9: 2523-9. Current Pharmaceutical Design, 2013, Vol. 19, No. 00 [127] [128] [129] [130] [131] [132] [133] [134] [135] [136] [137] [138] [139] [140] [141] [142] [143] [144] [145] [146] [147] [148] [149] [150] [151] 13 Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature 2007; 450: 1001-9. Christensen DE, Brzovic PS, Klevit RE. E2-BRCA1 RING interactions dictate synthesis of mono- or specific polyubiquitin chain linkages. Nat Struct Mol Biol 2007; 14: 941-8. Christensen DE, Klevit RE. Dynamic interactions of proteins in complex networks: identifying the complete set of interacting E2s for functional investigation of E3-dependent protein ubiquitination. FEBS J 2009; 276: 5381-9. Gardner RG, Shearer AG, Hampton RY. In vivo action of the HRD ubiquitin ligase complex: mechanisms of endoplasmic reticulum quality control and sterol regulation. Mol Cell Biol 2001; 21: 427691. Ballar P, Ors AU, Yang H, Fang S. Differential regulation of CFTRDeltaF508 degradation by ubiquitin ligases gp78 and Hrd1. Int J Biochem Cell Biol 2010; 42: 167-73. van Wijk SJ, de Vries SJ, Kemmeren P, et al. A comprehensive framework of E2-RING E3 interactions of the human ubiquitinproteasome system. Mol Syst Biol 2009; 5: 295. Markson G, Kiel C, Hyde R, et al. Analysis of the human E2 ubiquitin conjugating enzyme protein interaction network. Genome Res 2009; 19: 1905-11. Zheng N, Wang P, Jeffrey PD, Pavletich NP. Structure of a c-CblUbcH7 complex: RING domain function in ubiquitin-protein ligases. Cell 2000; 102: 533-9. Kar G, Keskin O, Nussinov R, Gursoy A. Human proteome-scale structural modeling of E2-E3 interactions exploiting interface motifs. J Proteome Res 2012; 11: 1196-207. Barabasi AL, Gulbahce N, Loscalzo J. Network medicine: a network-based approach to human disease. Nat Rev Genet 2011; 12: 56-68. Aloy P, Bottcher B, Ceulemans H, et al. Structure-based assembly of protein complexes in yeast. Science 2004; 303: 2026-9. Keskin O, Tsai CJ, Wolfson H, Nussinov R. A new, structurally nonredundant, diverse data set of protein-protein interfaces and its implications. Protein Sci 2004; 13: 1043-55. Tuncbag N, Gursoy A, Nussinov R, Keskin O. Predicting proteinprotein interactions on a proteome scale by matching evolutionary and structural similarities at interfaces using PRISM. Nat Protoc 2011; 6: 1341-54. Aytuna AS, Gursoy A, Keskin O. Prediction of protein-protein interactions by combining structure and sequence conservation in protein interfaces. Bioinformatics 2005; 21: 2850-5. Tuncbag N, Keskin O, Gursoy A. HotPoint: hot spot prediction server for protein interfaces. Nucleic Acids Res 2010; 38: W402-6. Ye Y, Rape M. Building ubiquitin chains: E2 enzymes at work. Nat Rev Mol Cell Biol 2009; 10: 755-64. Papaleo E, Ranzani V, Tripodi F, et al. An acidic loop and cognate phosphorylation sites define a molecular switch that modulates ubiquitin charging activity in Cdc34-like enzymes. PLoS Comput Biol 2011; 7: e1002056. Autore F, Pagano B, Fornili A, Rittinger K, Fraternali F. In silico phosphorylation of the autoinhibited form of p47(phox): insights into the mechanism of activation. Biophys J 2010; 99: 3716-25. Liu J, Nussinov R. Flexible cullins in cullin-RING E3 ligases allosterically regulate ubiquitination. J Biol Chem 2011; 286: 40934-42. Liu J, Nussinov R. Molecular dynamics reveal the essential role of linker motions in the function of cullin-RING E3 ligases. J Mol Biol 2010; 396: 1508-23. Patel S, George R, Autore F, et al. Molecular interactions of ASPP1 and ASPP2 with the p53 protein family and the apoptotic promoters PUMA and Bax. Nucleic Acids Res 2008; 36: 5139-51. Patel S, Bui TT, Drake AF, Fraternali F, Nikolova PV. The p73 DNA binding domain displays enhanced stability relative to its homologue, the tumor suppressor p53, and exhibits cooperative DNA binding. Biochemistry 2008; 47: 3235-44. Bruschweiler R. Protein dynamics: whispering within. Nat Chem 2011; 3: 665-6. Lakomek NA, Lange OF, Walter KF, et al. Residual dipolar couplings as a tool to study molecular recognition of ubiquitin. Biochem Soc Trans 2008; 36: 1433-7. Lange OF, Lakomek NA, Fares C, et al. Recognition dynamics up to microseconds revealed from an RDC-derived ubiquitin ensemble in solution. Science 2008; 320: 1471-5. 14 Current Pharmaceutical Design, 2013, Vol. 19, No. 00 [152] [153] [154] [155] [156] [157] [158] [159] [160] [161] [162] [163] [164] [165] [166] [167] [168] [169] [170] [171] [172] [173] [174] Wlodarski T, Zagrovic B. Conformational selection and induced fit mechanism underlie specificity in noncovalent interactions with ubiquitin. Proc Natl Acad Sci U S A 2009; 106: 19346-51. Long D, Bruschweiler R. In silico elucidation of the recognition dynamics of ubiquitin. PLoS Comput Biol 2011; 7: e1002035. Papaleo E, Casiraghi N, Arrigoni A, et al. Loop 7 of e2 enzymes: an ancestral conserved functional motif involved in the e2mediated steps of the ubiquitination cascade. PLoS One 2012; 7: e40786. Swaminathan G, Tsygankov AY. The Cbl family proteins: ring leaders in regulation of cell signaling. J Cell Physiol 2006; 209: 2143. Blake TJ, Shapiro M, Morse HC, 3rd, Langdon WY. The sequences of the human and mouse c-cbl proto-oncogenes show vcbl was generated by a large truncation encompassing a prolinerich domain and a leucine zipper-like motif. Oncogene 1991; 6: 653-7. Umebayashi K, Stenmark H, Yoshimori T. Ubc4/5 and c-Cbl continue to ubiquitinate EGF receptor after internalization to facilitate polyubiquitination and degradation. Mol Biol Cell 2008; 19: 3454-62. Makishima H, Cazzolli H, Szpurka H, et al. Mutations of e3 ubiquitin ligase cbl family members constitute a novel common pathogenic lesion in myeloid malignancies. J Clin Oncol 2009; 27: 6109-16. Joazeiro CA, Wing SS, Huang H, et al. The tyrosine kinase negative regulator c-Cbl as a RING-type, E2-dependent ubiquitinprotein ligase. Science 1999; 286: 309-12. Bertrand MJ, Milutinovic S, Dickson KM, et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell 2008; 30: 689-700. Varfolomeev E, Blankenship JW, Wayson SM, et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell 2007; 131: 669-81. Yang L, Cao Z, Yan H, Wood WC. Coexistence of high levels of apoptotic signaling and inhibitor of apoptosis proteins in human tumor cells: implication for cancer specific therapy. Cancer Res 2003; 63: 6815-24. Imoto I, Tsuda H, Hirasawa A, et al. Expression of cIAP1, a target for 11q22 amplification, correlates with resistance of cervical cancers to radiotherapy. Cancer Res 2002; 62: 4860-6. Varfolomeev E, Vucic D. (Un)expected roles of c-IAPs in apoptotic and NFkappaB signaling pathways. Cell Cycle 2008; 7: 1511-21. Flygare JA, Beresini M, Budha N, et al. Discovery of a potent small-molecule antagonist of inhibitor of apoptosis (IAP) proteins and clinical candidate for the treatment of cancer (GDC-0152). J Med Chem 2012; 55: 4101-13. Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 2000; 102: 33-42. Verhagen AM, Ekert PG, Pakusch M, et al. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 2000; 102: 43-53. Flygare JA, Fairbrother WJ. Small-molecule pan-IAP antagonists: a patent review. Expert Opin Ther Pat 2010; 20: 251-67. Al-Lazikani B, Banerji U, Workman P. Combinatorial drug therapy for cancer in the post-genomic era. Nat Biotechnol 2012; 30: 67992. Sato K, Hayami R, Wu W, et al. Nucleophosmin/B23 is a candidate substrate for the BRCA1-BARD1 ubiquitin ligase. J Biol Chem 2004; 279: 30919-22. Yu X, Fu S, Lai M, Baer R, Chen J. BRCA1 ubiquitinates its phosphorylation-dependent binding partner CtIP. Genes Dev 2006; 20: 1721-6. Starita LM, Horwitz AA, Keogh MC, et al. BRCA1/BARD1 ubiquitinate phosphorylated RNA polymerase II. J Biol Chem 2005; 280: 24498-505. Ma Y, Fan S, Hu C, et al. BRCA1 regulates acetylation and ubiquitination of estrogen receptor-alpha. Mol Endocrinol 2010; 24: 76-90. Esteller M, Silva JM, Dominguez G, et al. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J Natl Cancer Inst 2000; 92: 564-9. Kar et al. [175] [176] [177] [178] [179] [180] [181] [182] [183] [184] [185] [186] [187] [188] [189] [190] [191] [192] [193] [194] [195] [196] [197] [198] [199] Hogervorst FB, Nederlof PM, Gille JJ, et al. Large genomic deletions and duplications in the BRCA1 gene identified by a novel quantitative method. Cancer Res 2003; 63: 1449-53. Mishra A, Godavarthi SK, Jana NR. UBE3A/E6-AP regulates cell proliferation by promoting proteasomal degradation of p27. Neurobiol Dis 2009; 36: 26-34. Scheffner M. Ubiquitin, E6-AP, and their role in p53 inactivation. Pharmacol Ther 1998; 78: 129-39. Sanduja S, Kaza V, Dixon DA. The mRNA decay factor tristetraprolin (TTP) induces senescence in human papillomavirustransformed cervical cancer cells by targeting E6-AP ubiquitin ligase. Aging (Albany NY) 2009; 1: 803-17. Nachmias B, Ashhab Y, Ben-Yehuda D. The inhibitor of apoptosis protein family (IAPs): an emerging therapeutic target in cancer. Semin Cancer Biol 2004; 14: 231-43. Zhang Z, Wang H, Li M, et al. MDM2 is a negative regulator of p21WAF1/CIP1, independent of p53. J Biol Chem 2004; 279: 16000-6. Schlott T, Reimer S, Jahns A, et al. Point mutations and nucleotide insertions in the MDM2 zinc finger structure of human tumours. J Pathol 1997; 182: 54-61. Kim SS, Yoo NJ, Jeong EG, Kim MS, Lee SH. Expression of NEDD4-1, a PTEN regulator, in gastric and colorectal carcinomas. APMIS 2008; 116: 779-84. Dawson TM. Parkin and defective ubiquitination in Parkinson's disease. J Neural Transm Suppl 2006; 209-13. Read MA, Brownell JE, Gladysheva TB, et al. Nedd8 modification of cul-1 activates SCF(beta(TrCP))-dependent ubiquitination of IkappaBalpha. Mol Cell Biol 2000; 20: 2326-33. Oberg C, Li J, Pauley A, et al. The Notch intracellular domain is ubiquitinated and negatively regulated by the mammalian Sel-10 homolog. J Biol Chem 2001; 276: 35847-53. Yada M, Hatakeyama S, Kamura T, et al. Phosphorylationdependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J 2004; 23: 2116-25. Nateri AS, Riera-Sans L, Da Costa C, Behrens A. The ubiquitin ligase SCFFbw7 antagonizes apoptotic JNK signaling. Science 2004; 303: 1374-8. Koepp DM, Schaefer LK, Ye X, et al. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science 2001; 294: 173-7. Mao JH, Kim IJ, Wu D, et al. FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science 2008; 321: 1499-502. Akhoondi S, Sun D, von der Lehr N, et al. FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res 2007; 67: 9006-12. Frescas D, Pagano M. Deregulated proteolysis by the F-box proteins SKP2 and beta-TrCP: tipping the scales of cancer. Nat Rev Cancer 2008; 8: 438-49. Shapira M, Ben-Izhak O, Linn S, et al. The prognostic impact of the ubiquitin ligase subunits Skp2 and Cks1 in colorectal carcinoma. Cancer 2005; 103: 1336-46. Hershko DD. Oncogenic properties and prognostic implications of the ubiquitin ligase Skp2 in cancer. Cancer 2008; 112: 1415-24. Kavsak P, Rasmussen RK, Causing CG, et al. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol Cell 2000; 6: 1365-75. Rathmell WK, Chen S. VHL inactivation in renal cell carcinoma: implications for diagnosis, prognosis and treatment. Expert Rev Anticancer Ther 2008; 8: 63-73. Li Y, Zhou Z, Chen C. WW domain-containing E3 ubiquitin protein ligase 1 targets p63 transcription factor for ubiquitinmediated proteasomal degradation and regulates apoptosis. Cell Death Differ 2008; 15: 1941-51. Chen C, Zhou Z, Guo P, Dong JT. Proteasomal degradation of the KLF5 transcription factor through a ubiquitin-independent pathway. FEBS Lett 2007; 581: 1124-30. Li Y, Zhou Z, Alimandi M, Chen C. WW domain containing E3 ubiquitin protein ligase 1 targets the full-length ErbB4 for ubiquitin-mediated degradation in breast cancer. Oncogene 2009; 28: 2948-58. Neil JR, Tian M, Schiemann WP. X-linked inhibitor of apoptosis protein and its E3 ligase activity promote transforming growth factor-{beta}-mediated nuclear factor-{kappa}B activation during breast cancer progression. J Biol Chem 2009; 284: 21209-17. Emerging Role of the Ubiquitin-proteasome System [200] [201] [202] [203] [204] [205] Galban S, Duckett CS. XIAP as a ubiquitin ligase in cellular signaling. Cell Death Differ 2010; 17: 54-60. Carter BZ, Mak DH, Schober WD, et al. Simultaneous activation of p53 and inhibition of XIAP enhance the activation of apoptosis signaling pathways in AML. Blood 2010; 115: 306-14. Carter BZ, Mak DH, Schober WD, et al. Triptolide sensitizes AML cells to TRAIL-induced apoptosis via decrease of XIAP and p53mediated increase of DR5. Blood 2008; 111: 3742-50. Cummings J, Ranson M, Lacasse E, et al. Method validation and preliminary qualification of pharmacodynamic biomarkers employed to evaluate the clinical efficacy of an antisense compound (AEG35156) targeted to the X-linked inhibitor of apoptosis protein XIAP. Br J Cancer 2006; 95: 42-8. Schmitz R, Hansmann ML, Bohle V, et al. TNFAIP3 (A20) is a tumor suppressor gene in Hodgkin lymphoma and primary mediastinal B cell lymphoma. J Exp Med 2009; 206: 981-9. Priolo C, Tang D, Brahamandan M, et al. The isopeptidase USP2a protects human prostate cancer from apoptosis. Cancer Res 2006; 66: 8625-32. Received: October 2, 2012 Accepted: November 1, 2012 Current Pharmaceutical Design, 2013, Vol. 19, No. 00 [206] [207] [208] [209] [210] 15 Zhang D, Zaugg K, Mak TW, Elledge SJ. A role for the deubiquitinating enzyme USP28 in control of the DNA-damage response. Cell 2006; 126: 529-42. Faustrup H, Bekker-Jensen S, Bartek J, Lukas J, Mailand N. USP7 counteracts SCFbetaTrCP- but not APCCdh1-mediated proteolysis of Claspin. J Cell Biol 2009; 184: 13-9. van der Horst A, de Vries-Smits AM, Brenkman AB, et al. FOXO4 transcriptional activity is regulated by monoubiquitination and USP7/HAUSP. Nat Cell Biol 2006; 8: 1064-73. Mosesson Y, Mills GB, Yarden Y. Derailed endocytosis: an emerging feature of cancer. Nat Rev Cancer 2008; 8: 835-50. Deng S, Zhou H, Xiong R, et al. Over-expression of genes and proteins of ubiquitin specific peptidases (USPs) and proteasome subunits (PSs) in breast cancer tissue observed by the methods of RFDD-PCR and proteomics. Breast Cancer Res Treat 2007; 104: 21-30.