Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
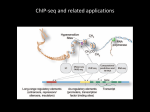
CHIP-SEQ: METHODS AND APPLICATIONS IN EPIGENOMIC STUDIES Claudia Angelini Istituto per le Applicazioni del Calcolo–CNR Napoli EMBO Practical course: Bioinformatics and Comparative Genome Analyses. May 15 Naples Outline Why study chromatin? Overview of a ChIP-seq experiment Overview on ChIP-seq data analysis Some (few) methods for peak calling Some (few) methods for peak annotation Discussion and conclusions Software Useful references Why study chromatin? Chromatin states are defined for a given time point and cell type Chromatin is the combination of DNA and proteins in eukaryotic cells Genome-wide mapping of protein-DNA interactions and epigenetic marks and their modifications is essential for a full understanding of transcriptional regulation and cell differentiation Chromatin states can influence transcription directly by altering the packing of the DNA Chromatin alterations are inheritable, but potentially reversible epigenetic drugs For a given (eukaryotic) genome there are hundreds or thousand of epigenomes depending on the chromatin states A scientific illustration of how epigenetic mechanisms can affect health http://commonfund.nih.gov/epigenomics/figure.aspx Chromatin Immunoprecipitation (ChIP) Chromatin Immunoprecipitation is a technique for assaying protein-DNA binding in vivo Antibodies are used to select specific proteins or nucleosomes which enriches for DNA-fragments that are bound to these proteins or nucleosomes Selected fragments can be either hybridized to a microarray (ChIP-chip) or sequenced on modern NGS platform (ChIPseq). Therefore, it is possible to profile the DNA bounds in vivo for specific transcription factors, modified histones, RNA Pol II Overview of a ChIP-seq experiment Park, nature review 2009 The DNA-binding protein is cross-linked to DNA in vivo by treating cells with formaldehyde the chromatin is sheared by sonication into small fragments, which are generally in the 200–600 bp range. an antibody specific to the protein of interest is used to immunoprecipitate the DNA–protein complex. the cross-links are reversed library are prepared according to the protocol and are usually subject to size selection the library is massively sequenced To account for experimental artifacts a control sample (i.e., Input) is (often) sequenced. Issues on experimental design Antibody quality Sample amount Control experiment (i.e., INPUT DNA) Depth of sequencing Samples’ number and amount of replicates Nowadays the use of a control experiment is a common practice. It is strongly suggested since it significantly improves the results. Saturation Analysis From experiment to “data” Park, nature review 2009 DNA fragments are sequenced from 5’ end to 3’ end the ChIP-seq protocols is strand specific and both strands are sequenced simultaneously The observed data are the “short reads” Short Read Binding site Fragment size Park , Nature Review 2009 Overview on ChIP-seq data analysis e.g, Input DNA 1) Identification of “enriched regions” (i.e., peak calling) 2) Interpretation of results, annotations, comparisons among samples and conditions Useful readings before starting with ChIP-seq From reads to coverage profile A typical ChIP-seq experiment produces from few millions to several tens of millions of short reads (depending on the experimental design and the organism under analysis) The first step in the analysis is the read mapping (i.e., the alignment to the reference genome) using standard tools such as Bowtie, BWA, etc Once the reads have been aligned the observed “profile” can be obtained and visualized Park, nature review 2009 Examples and Visualization Alignments can be uploaded in browsers such as UCSC Genome Browser or IGV etc… The “signal” appears in from of “peak” the problem is turned in detecting peaks The idea of Peak detection The peak detection is the most “critical” part of any ChIP-seq analysis. It consists in the identification of the genomic regions that were ‘enriched’ in the ChIP sample. In practice one has to find genomic regions that have produced a significantly higher number or reads (respect to the expected value estimated from the input) From Pepke et al, 2009 Few key ideas Compute the reads count at each position of the genome (either at bp resolution or within a given bin or [sliding] window) ChIP Determine if the “read count” is greater than expected Suitably combine pieces of small resolution (i.e., bp bins,…) into “regions” of enrichment Several issues & biases Reads are not uniformely distributed. Control (i.e., Input DNA) can be used for addressing such drawback GC-content PCR amplification Mappability ambiguity due repetitive regions Normalization is often necessary due to the different number of reads sequences in each library What to do? Methods for Peak detection There are several methods available These methods often require a careful choice/tuning of several parameters to obtain good results. Not always easy to use, often not well documented Some comparisons among methods (1) From Wilbanks & Facciotti 2010 Different methods can provide different results when applied to the same dataset. Comparing different methods is hard due to the lack of benchmark data-sets All methods rely on a set of parameters that need to be tuned accordingly to work best It is often necessary to use (or to try) several methods to the same dataset Methods are implemented in different programming languages. Some comparisons among methods (2) However the top (say 500-1000) peaks are usually consistent across methods, differences arise when looking for “marginal” peaks The use of control provides a significant improvement of the performance Deeper sequencing also improves performance (when control is available) Visualization is very important Despite the large number of methods available (more than 40!!), there is still space for improvement In the following few methods will be described more in details MACS (1) [Model-based Analysis of ChIP-Seq] It is one of the most popular approaches (also among the first to appear) It uses a Poisson distribution to model the reads and it empirically estimates the average fragment length using information from both strands It is implemented as standalone (freely available) software (in Python) It works either with only ChIP sample and with both ChIP and Control http://liulab.dfci.harvard.edu/MACS/ MACS (2) It estimates the average fragment size using a bimodal distribution on positive and negative strand (peak model), then shift the reads together by half of the distance toward the 3’ The number of reads are counted in each bin and the Poisson distribution (with local parameters estimated from the sample) is used to compute pvalues and identify the enriched regions. MACS (3) Local Poisson parameters are estimated when the control is available (otherwise uniform Poisson background is assumed) n 1 k e k k 0 k! X ~ Pois P X n 1 It reports either p-values and fold enrichment FDR is computed sample swapping Uniform background using To be estimated in window of size 1K,5K,10K from the control BayesPeak (1) it uses a fully Bayesian Hidden Markov model to detect enriched location in the genome All hyper-parameters are estimated using MCMC Enriched regions are detected on the basis of their posterior probability it works either with only ChIP sample and with both ChIP and Control It is implemented in the R package “BayesPeak” It is designed for the identification of transcription factor binding sites, but it has been also applied to Histone modifications such as H3K4me3 BayesPeak (2) Define the observed counts as Yt and Yt for window t, on the forward (+) and reverse (-) DNA strand. Assigns a state St to each region t such that S t 1 if there is a binding site or modification in that region causing fragment abundance, and S t 0 if not. PSt 1 1 | St 0 p PSt 1 1 | St 1 r Assuming among windows dependence adiacent Modeling the observed counts Equivalent to the use of Negative Binomial BayesPeak (3) The control sample can be used to estimate the hyper-parameters for the background Hyper-parameters are estimated via MCMC Inference is carried out on the posterior probability P(Z|Y) An “empirical” threshold T (default 0.5) have to be used on the posterior probability to select which regions are enriched Once P(Z|Y) is known, it is possible to compute P(S|Y) and several methods are available for combining bins into “regions” PICS (1) [Probabilistic Inference for ChIP-Seq] It uses a hierarchical Bayesian approach Instead of modeling read count, it models reads positions in positive and negative strands by using mixture-models It models the average fragment size as the distance of the two components of the mixture EM is used for estimating the hyper-parameters It uses information about mappability profiles (treated as missing values) It is implemented in the R package “PICS” It is designed for the identification of transcription factor binding sites. PICS (2) A filtering pre-processing step is applied and regions without a minimum number of reads in a windows are filtered out. Inference is carried out in each sub-regions independently (fitting mixture models) Parameters are obtained using EM the number of components in the mixture is chosen using BIC PICS (3) Reads that may not be aligned due to the repetitiveness of the genome are treated as missing (we do not know which read is missing but we may know where they be missing) Mappability profile (vector of 0-1 of length of the genome, where 1 means that a short read can be aligned to that position and 0 that it cannot be aligned) is used to handle repetitive regions Missing reads can handled with EM An enrichment score S() is defined to identify and rank a statistically significant sub-set of binding events FDR is estimated as function of the enrichment score after swapping the two samples Regions of no/or low mappability ChIP-seq: one “word” many “signal” Transcription Factor binding sites Histone modifications RNA polymerase II others From Wilbanks & Facciotti 2010 From Pepke et al, 2009 From TF to Histone modifications While there exists several methods able to detect TFs signatures along the genome, the identification of histone marks and their modifications is by far more challenging SICER Qeseq Rseg It contains also a comparative study Some comparisons among methods (3) Simulation Benchmarking publicly available ChIP-Seq datasets with qPCR validations ChIP-seq and repetitive regions (1) It is know that many TFs (or histone modifications) are associated to repetitive regions (also, 10-30% depending on the factors and the organism) Typical ChIP-seq analyis considers only uniquely mappable reads therefore introduce a bias in the possibility of studying the binding in repetitive regions Longer reads (and/or Paired-end protocols) reduce the problem, but it does not solve What to do ? Not yet a well accepted solution ChIP-seq and repetitive regions (2) 1) 2) Two emerging approaches Align the not uniquely reads on (known) repetitive regions and count the hits (per family or category) Assign the “multiple reads” using some euristic or some probabilistic model Good starting point From peaks to annotations and biological interpretation Once the “peaks” or the enriched regions have been identified they need to be annotated and biologically interpreted Peak annotation consists in relating the peaks’ position with all annotations and known genomic feature available (relevant for the problem under analysis) (TTS, genes, CpG, …). Biological interpretation depends on the problem under study. It includes motif dicovery and analysis, Gene ontology (Gene set enrichment-style analyses), pathway analysis, ….., etc. It may also require to combine information from several experiments and data integration. Data integration provides a more comprehensive complex picture of the biological system. It includes to relate peaks with expression levels of genes and SNPs and allele-specific binding ChIPpeakAnno R package for annotation It allows to integrate peak positions and annotations To visualize results using descriptive statistics To extract Goterms and test enrichment To visualize over representative sequences that can constitute a consensus rGADEM & MotIV R package motif finding RSAT [Regulatory Sequence Analysis Tools] http://rsat.ulb.ac.be/ A computational pipeline that discovers motifs in peak sequences, compares them with databases, exports putative binding sites for visualization in the UCSC genome browser and generates an extensive report suited for both naive and expert users GREAT [Genomic Regions Enrichment of Annotations Tool ] (1) GREAT supports direct enrichment analysis of both the human and mouse genomes. It integrates 20 separate ontologies containing biological knowledge about gene functions, phenotype and disease associations, regulatory and metabolic pathways, gene expression data, ….. http://great.stanford.edu/public/html/splash.php GREAT [Genomic Regions Enrichment of Annotations Tool ] (2) Two statistical tests are employed in order to establish the functional enrichment: Binomial test over genomic regions and Hypergeometric tests over genes Terms significant by both tests (B ∩ H) provide specific and accurate annotations supported by multiple genes and binding events. Terms significant by only the hypergeometric test (H\B) are general and often associated with genes of large regulatory domains, whereas terms significant by only the binomial test (B\H) cluster few genomic regions near only one or two genes annotated with the term Software availability http://seqanswers.com/wiki/Software On 21/04/2012 there are 558 “Bioinformatics Application”. Out of which about 47 are devoted to ChIP-seq Open Source & freely available Single stand-alone software (e.g. MACS, SICER,….) Specifics packages in general enviroments such as R (several in Bioconductor, see practical) Web resources (GALAXY, …) User friendly software with GUI (ChIPseeqer, Chipster,…) (General) Web-resoureces https://main.g2.bx.psu.edu/ Chipster http://chipster.csc.fi/ ChIPseeqer The software includes: Peak detection Gene-level annotation of peaks Pathways enrichment analysis Regulatory element analysis, using either a de novo approach, known or user-defined motifs Nongenic peak annotation (repeats, CpG islands, duplications) Conservation analysis Clustering analysis Visualization Integration and comparison across different ChIP-seq experiments http://physiology.med.cornell.edu/faculty/elemento/lab/chipseq.shtml Applications Studying disease processes Understanding basic regulatory mechanisms Studying cell differentiation ….. For what concerns comparative genome analyses, it is very important to study regulatory mechanisms across several species. A must to read for those interesting in comparative genome analyses Examples of ChIP-seq in comparative genome analysis Among several others Woo, Y.H, et al. Evolutionary Conservation of Histone Modifications in Mammals, Molecular Biology and Evolution, (2012) Hemberg, M. et al. Conservation of transcription factor binding events predicts gene expression across species, Nucl. Acids Res. (2011) 39 (16): 7092-7102. He, Q. et al. High conservation of transcription factor binding and evidence for combinatorial regulation across six Drosophila species. Nat. Genet. 43, 414–420 (2011). Schmidt, D. et al. Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science 328, 1036– 1040 (2010). Some other useful resources (1) COST Action BM1006: Next Generation Sequencing Data Analysis Network. http://www.seqahead.eu/ Training Schools and Short term scientific missions are available. Workshops are organized about twice times a year Some other useful resources (2) Officially started on 1/1/2012 References 11/05/2012 There are 590 papers with “Chip-seq” THANKS FOR YOUR ATTENTION ANY QUESTIONS?