Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
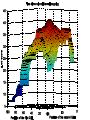
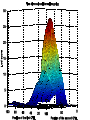
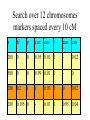
Modifying the Schwarz Bayesian Information Criterion to locate multiple interacting Quantitative Trait Loci 1. M.Bogdan, J.K.Ghosh and R.W.Doerge, Genetics 2004 167: 989-999. 2. M.Bogdan and R.W.Doerge “Mapping multiple interacting QTL by multidimensional genome searches’’ Xia- genotype of i-th individual at locus a Xia = 1/2 - individual is heterozygous at locus a Xia = -1/2 - individual is homozygous at locus a dab=10 cM - ρ (Xia, Xib) = 0.81 Data for QTL mapping Y1,...,Yn - vector of trait values for n backcross individuals X=[Xij], 1 ≤ i ≤ n, 1 ≤ j ≤ m - genotypes of m markers Standard methods of QTL mapping One QTL model (1) Yi Qi i , i N (0, ) 2 Qi (-1/2,1/2) - QTL genotype 1. Search over markers - fit model (1) at each marker and choose markers for which the likelihood exceeds a preestablished threshold value as candidate QTL locations. Interval mapping Lander and Botstein (1989) • Consider a fixed position between markers I i - state of flanking markers 1 1 1 1 1 1 1 1 I i , , , , , , , 2 2 2 2 2 2 2 2 1 pi P (Qi | I i ) easy to compute 2 Yi Qi i , i N (0, 2 ) 1 1 2 2 f (Yi | I i ) pi N ( , ) (1 pi ) N ( , ) 2 2 n L(Y | I ) f (Yi | I i ) i 1 1. Estimate μ, β, and σ by EM algorithm and compute the corresponding likelihood. 2. Repeat this procedure for a new possible QTL location. 3. Plot the resulting likelihoods as the function of assumed QTL position. • Problems with interval mapping a) Not able to distingush closely linked QTL b) Not able to detect epistatic QTL (involved only in interactions) • Solution Estimate the location of several QTL at once using multiple regression model (Kao et al. 1999) p Yi μ β jQij j1 r 1 j<l m γ jlQijQil ε i Problem : estimation of the number of additive and interaction terms p r j1 j1 Yi μ β jX ih j γ jX ik j X iu j ε i Xij - genotype of j-th marker average number of markers - (200,400) Bayesian Information Criterion • Choose the model which maximizes log L -1/2 k log n L – likelihood of the data for a given model k – number of parameters in the model n – sample size Broman (1997) and Broman and Speed (2002) – BIC overestimates QTL number How to modify BIC ? Mi – i-th linear model (specifies which markers are included in regression) θ = (μ, β1,..., βp, γ1,..., γr, σ) – vector of parameters for Mi fi(θ) – density of the prior distribution for θ π(i) – prior probability of Mi L(Y|θ) – likelihood of the data given the vector of paramers θ mi(Y) – likelihood of the data given the model Mi m i (Y) L(Y | θ)f i (θ)dθ P(Mi|Y) π(i)mi(Y) BIC neglects π(i) and uses asymptotic approximation log m i (Y) log L(Y, θ̂) 1/2(p r 2)log n neglecting π(i) = assigning the same prior probability to all models = assigning high prior probability to the event that there are many regressors Example : 200 markers 200 models with one additive term 200 =19 900 models with one interaction 2 or with two additive terms 200 = 9.05*1058 models with 100 additive terms 100 Idea: supplement BIC with a more realistic prior distribution π 1 ~ ˆ S (i ) log (i ) log L(Y , ) ( p(i ) r (i )) log n 2 n ˆ log L(Y , ) log RSS C (n) 2 RSS residual sum of squares from regression S (i ) n log RSS ( p(i ) r (i )) log n 2 log (i ) Choice of π (George and McCulloch, 1993) M – number of markers M(M 1) - number of potential interactions N 2 α - the probability that i-th additive term appears in the model ν - the probability that j-th interaction term appears in the model M- model with p additive terms and r interactions π(M)= αp νr(1-α)M-p (1-ν)N-r Prior distribution on the number of additive terms, p – Binomial (M,α) Prior distribution on the number of interactions, r – Binomial (N,ν) We choose 1 1 , l N and , u N l u log π(M)=C(M,N,l,u)-p log(l-1)-r log(u-1) S (i ) n log RSS ( p r ) log n 2 p log( l 1) 2r log( u 1) M E(p) , l N E(r) u Choice of l and u should depend on the prior knowledge on the number of QTL. Our choice – for the sample size 200 probability of wrongly detecting QTL (when there are none) ≈ 0.05 We keep E(p) and E(r) equal to 2.2 The choice is supported by theoretical bound on type I error based on Bonferoni inequality. S (i ) n log RSS ( p r ) log n 2p log( M / 2.2) 2r log( N / 2.2) Additional penalty similar to Risk Inflation Criterion of Foster and George (2k log t , where t is the total number of available regressors) and to the modification of BIC proposed by Siegmund (2004). Search over 12 chromosomes markers spaced every 10 cM n h2 p corr. extr 200 0 0 500 0 200 0.2 200 r corr extr 0.95 0.03 0 - 0.02 0 0.99 0.01 0 - 0 1 1 0.03 0 0 0.02 0.195 0 - 0.01 1 0.95 0.04 n h2 p corr extr r corr extr 200 0.55 0 - 0.02 3 2.88 0.08 200 0.5 7 5.06 0.26 0 - 0.09 500 0.5 7 6.99 0.14 0 - 0.03 200 0.43 12 2.39 0.31 0 - 0.03 500 0.43 12 9.68 0.47 0 - 0.02 200 0.71 12 9.53 0.75 0 - 0.02 200 0.53 2 1.95 0.04 5 2.11 0.11 500 0.53 2 2 0.03 5 3.47 0.08 • The criterion adjusts well to the number of available markers • For n = 200 the criterion detects almost all additive QTL with individual h2 =0.13 and interactions with h2 =0.2. • For n = 500 the criterion detects almost all additive QTL with individual h2 =0.06 and interactions with h2 =0.12. Bound for the type I error S1 the maximum of the criterion over all one dimensional models S0 = log L0 (Y / ˆ , ˆ ) the value of the criterion for the null model D - the number of terms chosen by our criterion P( D 0) P( S1 S0 ) S M i - the value of the criterion for a given one dimensional model S M i S0 if L(Y / ˆM i ) 2 log log n 2(log(l 1) or log(u 1)) L (Y / ˆ ) 0 0 P( S M i S0 ) 2 P( Z log n 2(log(l 1) or log(u 1))) where Z N (0,1) By Bonferoni inequality and the bound 2 1 x P(Z>x) exp( ) 2 2 x 2M 2N P( S1 S0 ) (l 1)C1 (l , n) (u 1)C2 (u, n) M N l , u 2.2 2.2 P( S1 S0 ) 4.4 1 1 2 n log n 2 log(l 1) log n 2 log(u 1) For n=200 and typical values of M this yields values in the range between 0.057 and 0.08.