Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
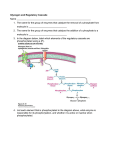
Journal Club Moslemi AR, Lindberg C, Nilsson J, Tajsharghi H, Andersson B, Oldfors A. Glycogenin-1 deficiency and inactivated priming of glycogen synthesis. N Engl J Med. 2010 Apr 1;362(13):1203-10. Lee IM, Djoussé L, Sesso HD, Wang L, Buring JE. Physical activity and weight gain prevention. JAMA. 2010 Mar 24;303(12):1173-9. 2010年4月8日 8:30-8:55 8階 医局 埼玉医科大学 総合医療センター 内分泌・糖尿病内科 Department of Endocrinology and Diabetes, Saitama Medical Center, Saitama Medical University 松田 昌文 Matsuda, Masafumi From the Department of Pathology, Institute of Biomedicine (A.-R.M., J.N., H.T., A.O.), Department of Neurology, Institute of Physiology and Neurological Sciences (C.L.), and Department of Cardiology, Institute of Medicine (B.A.), University of Gothenburg, Gothenburg, Sweden. N Engl J Med 2010;362:1203-10. Aim Glycogen, which serves as a major energy reserve in cells, is a large, branched polymer of glucose molecules. We describe a patient who had muscle weakness, associated with the depletion of glycogen in skeletal muscle, and cardiac arrhythmia, associated with the accumulation of abnormal storage material in the heart. Schematic illustration of glycogen synthesis. Unglucosylated dimers of apoglycogenin- 1 are autoglucosylated by an initial glucose-1-O-tyrosine linkage at Tyr195 (NM_004130), followed by addition of approximately 10 glucose molecules. This glycogenin oligosaccharide molecule constitutes the primer for synthesis of glycogen catalyzed by glycogen synthase and branching enzyme. Methods Analysis of genomic DNA Total DNA was extracted using the DNeasy Tissue Kit or the DNA Blood Mini Kit (Qiagen, Hilden, Germany). For PCR amplification we used primers that amplified the 8 exons of GYG1 (GenBank accession number NM_004130) with flanking intronic sequences (Supplementary Appendix, Table 2). The conditions for PCR were as follows: an initial denaturing step at 94°C for 3 min, followed by 35 cycles of: 94°C denaturing for 1 min, 57°C primer annealing for 1 min, 72°C primer extension for 1 min and a final extension step of 72°C for 10 min. PCR amplifications were performed with a GeneAmp PCR system 2700 (Applied Biosystems, Foster City, CA). Sequencing was performed using an ABI Prism 377 DNA sequencer and the Big Dye Terminator Kit v. 1. 1. (Applied Biosystems, Foster City, CA) and analyzed with MacVector® software 8.1.1. The coding sequences of GYS1, GBE1, GYG2 and PRKAG2 were amplified and sequenced in the same fashion. The missense mutation 248C>T in exon 3 of GYG1 eliminated a restriction site for endonuclease Tsp45I (New Englands Biolabs, Beverly, MA). For RFLP analysis of this mutation in genomic DNA, a 301-bp fragment was amplified using the forward and reverse exon 3 primers. Digestion was performed with 10 units of the enzyme at 37°C for two hours. The amplified 301-bp fragment of genomic DNA was digested into three fragments of 139, 154 and 8 bp, respectively, of normal DNA and into two fragments of 293 and 8 bp, respectively, of DNA carrying the 248C>T mutation. The fragments were then separated on a 2.5% agarose gel stained with GelStar® and visualized on a Dark Reader Blue light transilluminator (Clare Chemical Research, Dolores, CO). Analysis of cDNA For RFLP analysis of the GYG1 248C>T mutation in cDNA, total RNA was extracted using RNAqueous®-4PCR kit (Ambion, Austin, TX) and complementary DNA (cDNA) was synthesized using Ready-To-Go™ You-Prime First-Strand Beads (Amersham Biosciences, Buckinghamshire, UK). A 395-bp PCR product was amplified from GYG1 cDNA from the patient, parents and controls by using forward (F112) and reverse (R507) primers (GenBank accession number NM_004130). Restriction enzyme cleavage with Tsp45I and visualization of the results were performed as described above for genomic DNA. The amplified 395-bp fragment of cDNA was cleaved into two fragments of 243 and 152 bp, respectively, of wild type cDNA. Tissue culturing Muscle tissue specimens were cut into small pieces and seeded in flasks with Dulbeccos’ Modified Eagle’s Medium (DMEM), containing high glucose and glutamine levels and supplemented with 10% fetal calf serum and 1% penicillin. After 8-15 days the cells were detached enzymatically and plated in Petri dishes for proliferation. After expansion entailing 2-3 subcultures during two weeks, the cells were grown to 80% confluence and fusion was induced by switching to DMEM supplemented with 2% horse serum. The myoblasts and myotubes were then incubated overnight at 37°C in chamber slides. Western blot analysis For Western blot analysis, cultured muscle cells or cryostat sections of skeletal muscle and cardiac muscle, each ten μm thick, were homogenized in 100 μl Laemmli sample buffer supplemented with 5% ß-mercaptoethanol. To remove sugar residues from glycogenin, samples were treated with alpha-amylase (Sigma, St. Louis, MO), which hydrolyzes the internal α-1,4 glycosidic linkages of glycogen and autoglucosylated glycogenin. Total protein samples were loaded and separated on 3-8% Tris-Acetate or 10% Bis-Tris gels (Invitrogen, Carlsbad, CA) followed by electroblotting onto Invitrolon™ PVDF filters at 20 V for 10 minutes. The membrane was incubated with primary monoclonal mouse antihuman glycogenin-1 antibody (Abnova, Taipei City,Taiwan) 1:500, for one hour. Western Breeze™ (Invitrogen, Carlsbad, CA) was used for antibody detection. The same protocol was used with the following primary antibodies for Western blot analysis of other enzymes associated with glycogen synthesis: Anti-PRKAG2 (Atlas Antibodies, Stockholm, Sweden) 1:60, GBE1 (B01) (Abnova, Taipei City, Taiwan) 1:200 and Anti- Glycogen Synthase (Millipore, Temecula, CA) 1:1000. Recombinant protein expression and purification The full-length muscle isoform of glycogenin-1 was amplified from wild type and patient II:4’s muscle cDNA (with the 248C>T mutation) with the forward primer 5’- GGATCCATGACAGATCAGGCCTT-3’ and the reverse primer 5’CTCGAGCTGGAGGTAAGTGTCA-3’. The fragment was cloned into the pCDNA6/His Myc vector (Invitrogen, Carlsbad, USA) using BamHI and XhoI restriction enzymes. The final constructs were sequenced to confirm the expected sequences. Chinese-hamster-ovary (CHO K1) cells were cultured in Iscove’s Modified Dulbecco’s Medium (Lonza Biologicals, Basel, Switzerland), supplemented with 10% Fetal Bovine Serum (Lonza Biologicals, Basel, Switzerland) in 6-well culture plates (Falcon). Transfection of the vector was performed using Lipofectamine 2000 (Invitrogen, Carlsbad, USA), according to the manufacturer's instructions. The cells were harvested after 48 h and the His6-tagged recombinant glycogenin- 1 was purified using Ni-NTA spin columns (Qiagen, Hilden, Germany), in accordance with the manufacturer’s instructions. A 27-year-old man had dizziness and palpitations shortly after exercising, and emergency medical services were called. When the ambulance arrived, the patient was noted to have ventricular fibrillation, which was converted to sinus rhythm by cardiac defibrillation. After admission to the hospital, the patient had several short bursts of non sustained ventricular tachycardia. Cardiac magnetic resonance imaging in short axis view, showing a late enhancement area in mid-septum (arrow head) and an extensive area of late enhancement in the inferior wall (arrow). Courtesy Dr Carl Lamm, MD, PhD. Panel A demonstrates DNA sequence chromatograms from genomic DNA and cDNA with the identified mutations in exon 3, 248C>T, which results in the amino acid change Thr83Met, and in exon 5, 487delG, which results in a frame shift and a premature stop codon at amino acid position 167, Asp163ThrfsX5. Only the allele with the 248C>T mutation is expressed at the mRNA level, as determined by the cDNA sequence. Panels B (Normal control) and C (patient II:4) illustrate myoblasts and myotubes in tissue culture after staining with PAS reagent for glycogen. There is a deficiency of glycogen in the cells of patient II:4. Panel D illustrates results from RFLP analysis of cDNA from cultured myoblasts, using endonuclease Tsp45I that cleaves wild-type DNA, leaving DNA with the 248C>T (Thr83Met) mutation uncleaved. Only the mutated allele is expressed in the patient’s cultured myoblasts, whereas his father (I:2 in the pedigree) is heterozygous for the Thr83Met mutation. The myoblasts of the mother (I:1 of the pedigree), who does not carry this mutation, shows the same RFLP pattern as cultured myoblasts from a normal control. In Panel E the presence of glycogenin-1 in cultured myoblasts and myotubes is identified by Western blot analysis. In myoblasts from a normal control, a weak band of normal autoglucosylated glycogenin-1 can be identified. After alpha-amylase treatment to remove the polysaccharide chains from glycogen and glycogenin, a strong band of unglucosylated glycogenin-1 appears in the control. These glycogenin-1 molecules are approximately 1 kD smaller than the glycogenin-1 molecules identified without alpha-amylase treatment. In cultured myoblasts from patients with glycogen deficiency due to lack of glycogen synthase,11 normal autoglucosylated glycogenin-1 is identified without alphaamylase treatment. Alphaamylase treatment reduces the size of the molecule by approximately 1 kD. Analysis of glycogenin-1, without alpha-amylase treatment, in cultured myoblasts from patient II:4 demonstrates accumulation of glycogenin-1 with a molecular weight equal to that of normal glycogenin-1 after alpha-amylase treatment. The cultured cells of the father I:2 contain a larger proportion of unglucosylated glycogenin-1, compared to control myoblasts, which is consistent with the heterozygous expression of the Thr83Met mutation. Panel F illustrates the results of immunoblotting of three enzymes associated with glycogen synthesis. Patient II:4, who has the glycogenin-1 mutation, does not show any obvious upregulation of these enzymes. This is also the case for the patient with glycogen depletion due to glycogen synthase deficiency. Assessment of cardiac wall thickness and chamber dimensions by magnetic resonance imaging. Figure 1. Histochemical Images of Muscle-Biopsy Specimens. Muscle-biopsy specimens stained with periodic acid–Schiff reagent show strikingly less glycogen in the muscle fibers of the patient (Panel A) than in the fibers of the father (Panel B) and mother (Panel C), which contain normal levels. Panel D (with staining for myosin ATPase at pH 4.3) shows a marked predominance of dark, slow-twitch, oxidative (type 1) fibers in a specimen from the patient; Panel E shows fibers from a normal control. Panel F (with staining for succinate dehydrogenase) shows mitochondrial accumulation, especially in the subsarcolemmal region, in a specimen from the patient; Panel G shows a specimen from a normal control. The bars represent 30 μm. Figure 2. Histochemical and Ultrastructural Images of Myocardial-Biopsy Specimens. A myocardial-biopsy specimen from the patient’s right ventricle, shown in Panels A (hematoxylin and eosin), B (periodic acid–Schiff [PAS] reagent for glycogen), D, and E, is characterized by myocyte hypertrophy and large vacuoles (arrows) with PAS-positive material lacking the normal ultrastructural appearance of glycogen. Panel C (PAS reagent) shows evenly distributed glycogen in the intermyofibrillar network in a specimen from a normal control. The electron micrograph in Panel D shows one myocyte with a large vacuole (arrow), including scattered mitochondria (arrowheads); Panel E shows a mitochondrion (Mit) and the predominantly unstructured appearance of the storage material in the vacuole, which also contains lipid droplets (Lip) and small membrane-bound structures (arrowheads). An electron micrograph of a myocardialbiopsy specimen from a control subject, in Panel F, shows normal glycogen granules (arrows). Figure 4. Protein Analyses. Panel A shows the results of Western blot analysis of glycogenin-1 in skeletal muscle from a normal control, a patient with glycogen storage disease (Pompe’s disease due to alphaglucosidase deficiency), a patient with a lack of glycogen due to glycogen synthase deficiency,11 and the index patient in this study (Patient II-4). Without alpha-amylase treatment of the sample (−), glycogenin-1 was detectable only in the two persons who lacked glycogen. The size of the glycogenin-1 in these two persons differed by approximately 1 kD, since the glycogenin-1 was not autoglucosylated in the patient, who had the GYG1 Thr83Met mutation. Panel B shows the presence of glycogenin-1 in muscle identified by Western blot analyses performed with (+) and without (−) alpha-amylase treatment. In normal skeletal muscle, glycogenin-1 cannot be seen unless the sample is treated with alpha-amylase to remove the sugar residues from the huge glycogen molecules. This treatment will also hydrolyze the internal α-1,4glycosidic linkages between the autoglucosylated residues. In patients with deficient glycogen due to a lack of glycogen synthase,11 normal autoglucosylated glycogenin-1 can be shown without alpha-amylase treatment. Alpha-amylase treatment will reduce the size of the molecule by approximately 1 kD. Analysis of glycogenin-1 in the muscle of the patient (II-4), without alpha-amylase treatment, showed accumulation of glycogenin-1 with a molecular weight corresponding to that of normal glycogenin-1 after alpha-amylase treatment, demonstrating that the patient’s glycogenin-1 was unglucosylated. The muscle tissue from the father (I-2) contained a small proportion of unglucosylated glycogenin-1, visible without alpha-amylase treatment, which is compatible with the heterozygous expression of the allele carrying the Thr83Met mutation. In Panel C, Western blot analysis performed without treatment with alpha-amylase (−) shows accumulation of unglucosylated glycogenin-1 in cardiac tissue from the patient (II-4). In myocardium from control subjects, a small proportion of free autoglucosylated glycogenin-1 could be detected without alpha-amylase treatment, since the gel was loaded with a large amount of protein (as revealed by the myosin heavy-chain band). Panel D shows the results of Western blot analyses of human recombinant glycogenin-1. Wild-type and mutant (Thr83Met) GYG1 was expressed in Chinese-hamster-ovary cells. In this system, autoglucosylation will occur, but only of wild-type glycogenin-1, as shown by alpha-amylase treatment, which hydrolyzes any existing α-1,4-glycosidic linkages in autoglucosylated glycogenin-1. Treatment with alpha-amylase (+), as compared with no treatment (−), reduced the molecular weight of wild-type glycogenin-1 but not of Thr83Met mutant glycogenin-1, which demonstrates the inability of the mutant protein to autoglucosylate. The recombinant glycogenin-1 was tagged with 6-histidine for purification, resulting in an estimated molecular weight of approximately 40 kD. After examination and diagnostic testing, the patient underwent placement of an implantable cardioverter–defibrillator. Pharmacologic treatment was initiated with a β1-adrenergic-receptor blocker and an angiotensin-converting–enzyme inhibitor. The patient has had no further cardiac arrhythmias and no clinical signs or symptoms of heart failure. At follow-up 1 year after the acute episode, his physical capacity was categorized as New York Heart Association functional class I or II. Results The skeletal muscle showed a marked predominance of slow-twitch, oxidative muscle fibers and mitochondrial proliferation. Western blotting showed the presence of unglucosylated glycogenin-1 in the muscle and heart. Sequencing of the glycogenin-1 gene, GYG1, revealed a nonsense mutation in one allele and a missense mutation, Thr83Met, in the other. Conclusion In summary, we have described a metabolic disease that is due to a deficiency of glycogenin-1. The disease affects the priming of glycogen synthesis and results in glycogen depletion and accumulation of abnormal storage material in the heart. The missense mutation resulted in inactivation of the autoglucosylation of glycogenin-1 that is necessary for the priming of glycogen synthesis in muscle. Message 筋肉のグリコーゲン合成系の代謝異常で,神経, 循環器,筋肉疾患となる。 Division of Preventive Medicine (Drs Lee, Sesso, Wang, and Buring) and Aging (Drs Djousse´ , Sesso, and Buring), Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School; Department of Epidemiology, Harvard School of Public Health (Drs Lee and Buring); Department of Ambulatory Care and Prevention, Harvard Medical School (Dr Buring), and Massachusetts Veterans Epidemiology and Research Information Center, Boston Veterans Affairs Healthcare System (Dr Djousse´ ) Boston, Massachusetts. JAMA. 2010;303(12):1173-1179 MET: metabolic equivalent Physical Activity MET Light Intensity Activities <3 sleeping 0.9 watching television 1.0 writing, desk work, typing 1.8 walking, less than 2.0 mph (3.2 km/h), level ground, strolling, very slow 2.0 Moderate Intensity Activities 3 to 6 bicycling, stationary, 50 watts, very light effort 3.0 calisthenics, home exercise, light or moderate effort, general 3.5 bicycling, <10 mph (16 km/h), leisure, to work or for pleasure 4.0 bicycling, stationary, 100 watts, light effort 5.5 Vigorous Intensity Activities >6 jogging, general 7.0 calisthenics (e.g. pushups, situps, pullups,jumping jacks), heavy, vigorous effort 8.0 running jogging, in place 8.0 Background Context: The amount of physical activity needed to prevent long-term weight gain is unclear. In 2008, federal guidelines recommended at least 150 minutes per week (7.5 metabolic equivalent [MET] hours per week) of moderate-intensity activity for “substantial health benefits.” Objective: To examine the association of different amounts of physical activity with long-term weight changes among women consuming a usual diet. Method Design, Setting, and Participants: A prospective cohort study involving 34 079 healthy US women (mean age, 54.2 years) from 1992-2007. At baseline and months 36, 72, 96, 120, 144, and 156, women reported their physical activity and body weight. Women were classified as expending less than 7.5, 7.5 to less than 21, and 21 or more MET hours per week of activity at each time. Repeated measures regression prospectively examined physical activity and weight change over intervals averaging 3 years. Main Outcome Measure: Change in weight. Results Women gained a mean of 2.6 kg throughout the study. A multivariate analysis comparing women expending 21 or more MET hours per week with those expending from 7.5 to less than 21 MET hours per week showed that the latter group gained a mean (SD) 0.11 kg (0.04 kg; P=.003) over a mean interval of 3 years, and those expending less than 7.5 MET hours per week gained 0.12 kg (0.04; P=.002). There was a significant interaction with body mass index (BMI), such that there was an inverse dose-response relation between activity levels and weight gain among women with a BMI of less than 25 (P for trend=.001) but no relation among women with a BMI from 25 to 29.9 (P for trend=.56) or with a BMI of 30.0 or higher (P for trend=.50). A total of 4540 women (13.3%) with a BMI lower than 25 at study start successfully maintained their weight by gaining less than 2.3 kg throughout. Their mean activity level over the study was 21.5 MET hours per week (~60 minutes a day of moderate intensity activity). Conclusion Among women consuming a usual diet, physical activity was associated with less weight gain only among women whose BMI was lower than 25. Women successful in maintaining normal weight and gaining fewer than 2.3 kg over 13 years averaged approximately 60 minutes a day of moderate-intensity activity throughout the study. Message 肥満度が高いと運動はあまり減量には効果はない。 体重維持には 1日60分の運動が必要!