Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
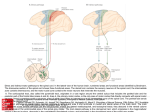
21 April 2017 EMA/250453/2017 Media and Public Relations Press release First medicine for spinal muscular atrophy Orphan medicine Spinraza recommended by CHMP under accelerated assessment The European Medicines Agency (EMA) has recommended granting a marketing authorisation in the European Union (EU) for Spinraza (nusinersen) to treat patients with spinal muscular atrophy (SMA), a rare and often fatal genetic disease that causes muscle weakness and progressive loss of movement. Spinraza is to be given by lumbar puncture injection into the fluid surrounding the spinal cord once every four months. There is currently no approved therapy in the EU for the treatment of spinal muscular atrophy. Patients receive supportive treatment to help them and their families cope with the symptoms of the disease. This includes chest physiotherapy and physical aids to support muscular function, and mechanical ventilators to help with breathing. There is therefore a significant unmet medical need for these patients. Spinal muscular atrophy is an inherited disease usually diagnosed in the first year of life that affects the motor neurons (nerves from the brain and spinal cord that control muscle movements). Patients with the disease lack a protein called ‘survival motor neuron’ (SMN), which is essential for the normal functioning and survival of motor neurons. Without this protein, the motor neurons deteriorate and eventually die. This causes the muscles to fall into disuse, leading to muscle wasting (atrophy) and weakness. The SMN protein is made by two genes, the SMN1 and SMN2 genes. Most patients with spinal muscular atrophy lack the SMN1 gene but have the SMN2 gene, which mostly produces a ‘short’ SMN protein which cannot work properly on its own. Spinraza is an ‘anti-sense oligonucleotide’ medicine. It is expected to make the SMN2 gene produce adequate levels of the SMN protein of normal length, thereby increasing the survival of motor neurons. The recommendation from the CHMP is based on the results of one completed clinical trial and a number of ongoing trials in patients with spinal muscular atrophy with different stages of disease severity. These included patients with infantile-onset, as well as later childhood-onset of the disease, as well as patients in the pre-symptomatic phase. 30 Churchill Place ● Canary Wharf ● London E14 5EU ● United Kingdom Telephone +44 (0)20 3660 8427 Facsimile +44 (0)20 3660 5555 E-mail [email protected] Website www.ema.europa.eu An agency of the European Union The clinical trial providing the main body of data for the assessment was conducted in 121 patients with infantile-onset who were randomised to receive an injection of Spinraza into the fluid surrounding the spinal cord, or undergo a mock procedure without an injection (a skin prick). The trial assessed the percentage of patients who had a pre-defined level of improvement (responders) in their motor milestones such as head control, rolling, sitting, crawling, standing and walking. Fifty one per cent of patients met the criteria as responders to treatment with Spinraza, compared to none receiving the mock injection (controls). Sixteen patients (22%) achieved full head control, six patients (8%) achieved independent sitting, and one patient (1%) achieved standing with support, whereas no subjects in the control group achieved any of these milestones. In the same trial, the risk of death or permanent ventilation was 47% less in patients treated with Spinraza. Unfortunately, 49% of the patients treated with Spinraza in the trial were not considered to be motor milestone responders. These effects are considered to be of considerable clinical importance as patients with infantile-onset spinal muscular atrophy typically fail to achieve independent sitting, do not see any improvement in their motor skills beyond those present at the time of diagnosis, and usually die within the first two years without intensive supportive care, as a result of progressive muscle weakness. Additional data were obtained from a separate, ongoing clinical trial conducted in 126 patients with later childhood-onset spinal muscular atrophy who were randomised to receive Spinraza or undergo a mock procedure without an injection. The trial compared the motor skills of the patients assessed at 15 months after treatment with those assessed when they entered the trial, using the Hammersmith Functional Motor Scale – Expanded (HFMSE). The group of patients treated with Spinraza improved by four points on the scale at 15 months, while controls declined by 1.9 points. This is an important outcome in patients who typically experience a gradual decline in their motor ability over time. The results obtained in these two randomised, double-blind, sham controlled clinical trials were supported by results from clinical trials with no comparators. In one of these trials, infants genetically diagnosed with the disease, but who did not yet have symptoms, received injections of Spinraza and seemed to achieve motor milestones comparable to normal development. Because patients treated with Spinraza have not yet been followed for a long period of time, it is not yet known whether the effects of Spinraza will be maintained in the longer term, or whether Spinraza may be able to provide a cure in some of the SMA patients. More information on these aspects will become available with time. Data are very limited in the milder forms of SMA associated with later age of onset and less severe outcomes. Nevertheless an effect of Spinraza can be assumed in these patients because it works in the same way as in more severely affected patients. Data will be collected post approval to confirm that this is the case. The most common side effects found in participants in the clinical trials for Spinraza were upper respiratory infection, lower respiratory infection and constipation. Spinraza must be administered by healthcare professionals experienced in doing lumbar punctures. Spinal muscular atrophy is a rare disease and patients with the condition have a significant unmet medical need. The Agency has a number of mechanisms to encourage the development of medicines in such situations. Spinraza received an orphan designation in 2012, with the consequent incentives including free scientific advice on the clinical and non-clinical aspects of the medicine’s dossier. Once the application was made, the Agency reviewed it under its accelerated assessment programme, designed to facilitate access to medicines that meet an unmet medical need. First medicine for spinal muscular atrophy Page 2/3 In November 2016, the Agency hosted a workshop bringing together patients and academics to progress research into spinal muscular atrophy and facilitate the development of new therapies. The opinion adopted by the CHMP at its April 2017 meeting is an intermediary step on Spinraza’s path to patient access. The CHMP opinion will now be sent to the European Commission for the adoption of a decision on EU-wide marketing authorisation. Once a marketing authorisation has been granted, a decision about price and reimbursement will then take place at the level of each Member State considering the potential role/use of this medicine in the context of the national health system of that country. Notes 1. This press release, together with all related documents, is available on the Agency's website. 2. The applicant for Spinraza is Biogen Idec Ltd. 3. Following this positive CHMP opinion, the Committee for Orphan Medicinal Products (COMP) will assess whether the orphan designation should be maintained. 4. More information on the work of the European Medicines Agency can be found on its website: www.ema.europa.eu Contact our press officer Monika Benstetter Tel. +44 (0)20 3660 8427 E-mail: [email protected] Follow us on Twitter @EMA_News First medicine for spinal muscular atrophy Page 3/3