Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Discovery and development of tubulin inhibitors wikipedia , lookup
Pharmaceutical marketing wikipedia , lookup
Specialty drugs in the United States wikipedia , lookup
Discovery and development of proton pump inhibitors wikipedia , lookup
Polysubstance dependence wikipedia , lookup
Compounding wikipedia , lookup
Orphan drug wikipedia , lookup
Plateau principle wikipedia , lookup
Drug design wikipedia , lookup
Drug discovery wikipedia , lookup
Neuropharmacology wikipedia , lookup
Pharmacogenomics wikipedia , lookup
Psychopharmacology wikipedia , lookup
Pharmacognosy wikipedia , lookup
Neuropsychopharmacology wikipedia , lookup
Pharmaceutical industry wikipedia , lookup
Prescription costs wikipedia , lookup
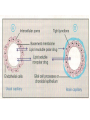
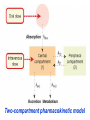
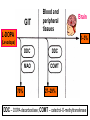
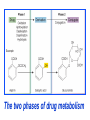
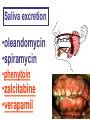
GENERAL PHARMACOKINETICS Assoc. Prof. I. Lambev Medical University of Sofia www.medpharm-sofia.eu (Abstract) Pharmacokinetics – how does the human body act on the drugs? Pharmacokinetics is the quantitative study of drug movement in, through and out of the body. Intensity of effect is related to the concentration of the drug at the site of action, which depends on its pharmacokinetic properties. Pharmacokinetic properties of the drug determine the route(s) of administration, dose, latency of onset, time of peak action, duration of action and frequency of drug administration. All pharmacokinetics processes involve transport of the drug across biological lipid membrane. Passive diffusion Filtration through lipid Carrier transport Passive transport - Passive diffusion - Filtration Specialized transport - Carrier transport Active transport Facilitated diffusion - Pinocytosis, etc. Convection Passive (simple) diffusion The lipid soluble unionized drug diffuse across the lipid biomembrane in the direction of their concentration gradient. It does not need energy. Most drugs are week electrolytes. Their ionization is pH dependent. The ionization of a week acid (AH) is given by the equation of Henderson–Hasselbalch: [AH] pKa = pH + log10 -----[A ] pKa is the negative logarithm of acidic dissociation constant of the week electrolyte. If the concentration of unionized drug [AH] is equal to concentration of ionized drug [A-], then [AH] --------- = 1 [A-] since log 1 is 0, under this condition pH = pKa In this case the molecules of drugs are 50% ionized. For a week base: pKb = pH + [BH+] log10 -----[B] Filtration Filtration is passage of a drug through aqueous pores in the membrane through paracelullar spaces. The moving force is hydrostatic or osmotic pressure. Lipid insoluble drugs cross the biomembrane by filtration only if their molecular size is smaller than the diameter of the enlarged aqueous pores. The filtration has an importance mainly at the level of renal glomerulus, where the size of capillaries have large pores (40 Å) and most drugs (even albumin) can filtrate. The brain capillary pores have small size. Carrier transport – by combination with a carrier molecule which acts as a ferry-boat across the lipid region of the membrane. Carrier transport is saturable and competitively inhibited by analogues which utilize the same carrier. a) Active transport is a movement against the concentration gradient. It needs energy and is inhibited by metabolic poisons. Levodopa and methyldopa are actively absorbed from the gut by aromatic amino acid transport. b) Facilitated diffusion. This proceeds more rapidly than passive (simple) diffusion and translocates even nondiffusible substrates, but along their concentration gradient, therefore, does not need energy. Example: Facilitated transport of glucose. Pinocytosis involves the invagination of a part of the cell membrane and trapping within the cell of a small vesicle containing extracellular constituents. The vesicle contents can than be released within the cell, or extruded from the other side of the cell. Pinocytosis is important for the transport of some macromolecules (e.g. insulin through BBB). I. ABSORPTION It is the passage of drug from the site of administration into the circulation. Aqueous solubility. Drugs given in solid form must dissolve in the aqueous biophase before they are absorbed. For poorly water soluble drugs (aspirin, griseofulvin) the rate of dissolution governs the rate of absorption. If a drug is given as water solution, it is absorbed faster than the same given in solid form or as a oily solution. Concentration. Passive transport depends on the concentration gradient. A drug given as concentrated solution is absorbed faster than dilute solution. Area of absorbing surface. If the area is larger, the absorption is faster. Vascularity of absorbing surface. Blood circulation removes the drug from the site of absorption and maintains concentration gradient across the membrane. Increased blood flow hastens drug absorption. Route of administration affects drug absorption, because each route has its own peculiarities. Oral application. Unionized lipid soluble drugs (e.g. ethanol) are readily absorbed from GIT. Acid drugs (aspirin, barbiturates, etc.) are predominantly unionized in the acid gastric juice and are absorbed from the stomach. Acid drugs absorption from the stomach is slower, because the mucosa is thick, covered with mucus and the surface is small. Basic drugs (e.g. atropine, morphine, etc.) are largely ionized and are absorbed only from the duodenum. Presence of food dilutes the drug and retards absorption. Certain drugs form poorly absorbed complexes with food constituents, e.g. tetracyclines with calcium present in milk. Food delays gastric emptying.Most drugs are absorbed better if taken on an empty stomach. Highly ionized drugs, e.g. amikacin, gentamicin, neostigmine, are poorly absorbed when given orally. Certain drugs are degraded in the GIT, e.g. penicillin G by acid, insulin by peptidases, and are ineffective orally. Enteric coated tablets (having acid resistant coating) and sustained released preparations can be used to overcome acid ability, gastric irritancy and brief duration of action. Intestinal absorption: 2+ - duodenum (B1, Fe ) - ileum (B12, A, D, E, K) - large intestine + + (water, Na , Cl , K ) Drugs can also alter absorption by gut wall effect: altering motility (atropine, amitriptyline, pethidine, methoclopramide) or causing mucosal damage (neomycin, methotrexate, reserpine, vinblastine). Alteration of gut flora by antibiotics may disrupt the enterohepatic recirculation of oral contraceptives and digoxin. S.c. and i.m. application By these routes the drug is deposited in the vicinity of the capillaries. Lipid soluble drugs pass readily across the whole surface of the capillary endothelium, but very large molecules are absorbed through lymphatics. Many drugs not absorbed orally are absorbed parenterally. Absorption from s.c. site is slower than that from i.m. site, but both are generally faster and more predictable than p.o. absorption. Application of heat and muscular exercise accelerate drug absorption by increasing blood flow. Application of vasoconstrictors (e.g. adrenaline) retard absorption. Many depot preparations (preparations with a long action), such as benzatine benzylpenicillin and protamine zinc insulin can be given by these routes. Topical applications (skin, cornea, mucous membranes) Systemic absorption depends on lipid solubility. Only a few drugs significantly penetrate intact skin. Nitroglycerine, hyoscine (scopolamine) and estradiol have been used in this manner. Glucocorticosteroids (GCS) applied over extensive areas can produce systemic effects and pituitary-adrenal suppression. Cornea is permeable to lipid soluble, unionized physostigmine but not to highly ionized neostigmine. Similarly, the mucous membrane of the mouth, rectum and vagina absorb lipophilic drugs, e.g. estrogen cream applied intravaginally has produced gynecomastia in the male partner. Bioavailability refers to the rate and extent of absorption of a drug from dosage form as determined by its concentration-time curve in blood or by its excretion in urine. It is a measure of the fraction (F) of administered dose of a drug that reaches the systemic circulation in the unchanged form. Bioavailability of a drug injected i.v. is 100%, but is frequently lower after oral ingestion, because: a) The drug may incompetely absorb b) The absorbed drug may undergo first pass metabolism in intestinal wall and/or liver, or be excreted in bile. Plasma concentration (mcg/ml) AUC – area under the curve F – bioavailability AUC p.o. F = ------------ x 100% AUC i.v. (i.v. application) (p.o. application) 0 5 Time (h) 10 15 Plasma concentration time curves of the three preparations of a drug which contain the same amount. Formulation B is more slowly absorbed than A and may not produce therapeutic effect. Formulation C is absorbed to a lesser extent (it has lower bioavailability). II. DISTRIBUTION In studying the pharmacokinetics biosystems conditionally divided into separate parts – compartment (or phases). They are virtual spaces in which the drug is evenly distributed. They are distinguished each other in the volume of distribution and invasion (penetration) and evazionnite (release) rate constants. The distribution of the drugs is a dynamic process, during which they pass from the central (plasma) compartment in the tissue to reach steady state (steady state – ss). It depends on the mode of administration and the pK of the drug, its ability for binding to plasma protein, pH of the medium, organ perfusion. The number of compartments is determined for each drug according to the experimental data measured concentrations at different moments in the blood, urine and body fluids. Two-compartment pharmacokinetic model Body fluid compartments The total body water as a percentage of body mass varies from 50% to 70%, being rather less in women than in man. Body water is distributed into the following main compartments: 1. plasma (5% of body mass) 2. intestinal fluid (16%) 3. intracellular fluid (35%) 4. transcellular fluid (2%) 5. fat (20%) Apparent volume of distribution (Vd) It is accept that the body behaves as a single homogeneous compartment with volume (Vd) in which the drug gets immediately distributed: Dose administered Vd = ----------------------------Plasma concentration Drugs extensively bound to plasma proteins are largely restricted to the vascular compartment and have low Vd (e.g. warfarin – 99% bound and its Vd is 0,1 L/kg). Drugs sequestrated in other tissues may have Vd much more than the total body water or even body mass, e.g. digoxin (6 L/kg) and propranolol (3 to 4 L/kg) because most of the drug is present in other tissues, and the plasma concentration is low. Therefore, in case of poisoning, drugs with large Vd are not easily removed by haemodialysis. Redistribution. Highly lipid soluble drugs given i.v. or by inhalation get distributed to organs with high blood flow (brain, heart, kidney, liver). Later they get distributed to less vascular tissues (muscles and fat) and the drug-plasma concentrations falls. The greater lipid solubility of the drug hastens its redistribution. Anaesthetic action of thiopentone (thiopental) is terminated in few minutes due to redistribution. However, when the same drug is given repeatedly or continuously over long periods the low perfusion high capacity sites get progressively filled up and the drug becomes longer acting. Thiopental (thiopentone) -redistribution in muscle and fat (long postnarcotic sleep) Blood brain barrier (BBB): includes the capillary endothelial cells (which have tight junctions and lack large intracellular pores) and an investment of glial tissue, over the capillaries. A similar barrier is loctated in the choroid plexus. BBB is lipid and limits the entry of non-lipid soluble drugs (amikacin, gentamicin, neostigmine etc.). Only lipid soluble unionized drugs penetrate and have action on the CNS. Efflux carriers like P-gp (glycoprotein) present in brain capillary endothelial cells (also in intestinal mucosal, renal tubular, hepatic canicular, placental, and testicular cells) extrude drugs that enter the brain by other processes. Inflammation of the meninges of the brain increases permeability of the BBB. Dopamine (DA) does not enter the brain, but its precursor levodopa does. This is used later in parkinsonism. GIT L-DOPA Blood and peripheral tissues Brain 1–3% (Levodopa) 70% DDC DDC МАО COMT 27–29% DDC – DOPA-decarboxilase; COMT – catechol-О-methyltransferase Placental barrier. Placental membranes are lipid and allow free passage of lipophilic drug, while restricting hydrophilic drugs. The placental P-gp also serves to limit foetal exposure to maternally administered drugs. However restricted amounts of nonlipid soluble drugs, when present in high concentration or for long periods in maternal circulation, gain access to the foetus. Thus, it is an incomplete barrier and many drugs, taken by the mother, can affect the foetus or the newborn. Penicillins, azithromycin, and erythromycin do not affect the foetus and can be used during the pregnancy. Plasma protein binding (PPB). Most drugs possess physicochemical affinity for plasma proteins. Acidic drugs bind to plasma albumin and basic drugs to α1-glycoprotein. Extent of binding depends on the individual compound. Increasing the concentration of a drug can progressively saturate the binding sites. The clinical significant implications of PPB are: a) Highly PPB drugs are largely restricted to the vascular compartment and tend to have lower Vd. b) The PPB fraction is not available for action. c) There is an equilibration between the PPB fraction of the drug and the free molecules of the drug. d) The drugs with high physicochemical affinity for plasma proteins (e.g. aspirin, sulfonamides, chloramphenicol) can replace the other drugs (e.g. acenocoumarol, warfarin) or endogenous compounds (bilirubin) with lower affinity. e) High degree of protein binding makes the drug longacting, because bound fraction is not available for metabolism, unless it is actively excreted by the liver or kidney tubules. f) Generally expressed plasma concentrations of the drug refer to bound as well as free drug. g) In hypoalbuminemia, binding may be reduced and high concentration of free drug may be attained (e.g. phenytoin). Tissue storage. Drugs may also accumulate in specific organs or get bound to specific tissue constituents, e.g.: Heart and skeletal muscles – digoxin (to muscle proteins) Liver – chloroquine, tetracyclines, digoxin Kidney – digoxin, chloroquine Thyroid gland – iodine Brain – chlorpromazine, isoniazid, acetazolamide Retina – chloroquine (to nucleoproteins) Iris – ephedrine, atropine (to melanin) Bones and teeth – tetracyclines, heavy metals (to mucopolysaccharide of connective tissue) Adipose tissues – thiopental, ether, minocycline, DDT III. METABOLISM (BIOTRANSFORMATION) Metabolism includes chemical alteration of the drugs in the body. Most hydrophilic drugs (amikacin, gentamycin, neostigmine, mannitol) are not biotransformated and are excreted unchanged. The mechanism to metabolize drugs is developed to protect the body from toxins. The primary site for drug metabolism is the liver, other sites are the kidney, intestine, lungs, and plasma. Metabolism of drugs may lead to the following: a) Inactivation. Most drugs and their active metabolites are converted to less active or inactive metabolites, e.g. phenobarbital, morphine, propranolol, etc. b) Active metabolite from an active drug. Many drugs are converted to one or more active metabolites (e.g. diazepam, amitriptyline). c) Activation of inactive drug. Few drugs (so called prodrugs) are inactive as such. They need conversion in the body to one or more active metabolites (e.g. levodopa, benfothiamine, enalapril, perindopril). The prodrug may offer advantages: their active forms may be more stable; they can have better bioavailability (e.g. benfothiamine), or other desirable pharmacokinetic properties or less side effects and toxicity. Biotransformation reactions can be classified into two phases: I (no synthetic) and II (synthetic, conjugation). Phase I (no synthetic reactions) a) Oxidation is the most important drug metabolizing reaction. Various oxidation reactions are hydroxylation; oxygenation at C-, N- or S-atoms; N or 0-dealkylation, oxidative deamination, etc. Oxidative reactions are mostly carried out by a group of monooxygenases in the liver, which in the final step involve cytochrome P450 reductase and O2. There are more than 200 cytochrome P450 isoenzymes, differing in their affinity for various substances (drugs). They are grouped into > 20 families. CYP 3A4/5 carry out biotransformation of the largest number (≈ 50%) of drugs. In addition to the liver, these isoforms are expressed in the intestine (responsible for first pass metabolism at this site) and the kidney too. Inhibition of CYP 3A4 by erythromycin, clarithromycin, ketoconzole, itraconazole, verapamil, diltiazem, and a constituent of grape fruit juice are responsible for unwanted interaction with terfenadine. Rifampicin, phenytoin, carbmazepine, phenobarbital are inducers of the CYP 3A4. Substrates: Cumarins CYP2B6 CYP2A6 <5% Mephenytoin Omeprazole CYP2C19 <5% Tolbutamide Warfarin Phenytoin CYP2C8/9/18 ~20% Midazolam Nifedipine Erythromycin Cyclosporine CYP3A4/5 (30–50%) Caffeine Theophylline Tacrine CYP1A2 ~15% Chlorzoxazone CYP2E1 ~10% Dextrometorphan Debrisoquine CYP2D6 <5% CYP1A1 Inhibitors: Methoxsalen Fluconazole Sulphaphenazole Ketoconazole Gestodene Furafylline Fluvoxamine Tetrahydro- Quinidine furane Inducers: Phenobarbital Phenobarbital Phenobarbital Rifampicin Rifampicin Phenobarbital Rifampicin Dexamethasone Carbamazepine Omeprazole Nicotine Ethanol Isoniazid Barbiturates, phenothiazines, paracetamol, streroids, phenytoin, benzodiazepines, theophyllin and many other drugs are oxydaized by CYP450. Some other drugs (adrenaline, mercaptopurine) and ethanol are oxidized by mitochondrial or cytoplasmic enzymes. b) Reduction. This reaction is conversed of oxidation and involves CYP450 enzymes working in the opposite direction. Drugs, primarily reduced, are chloramphenicol, halothane. c) Hydrolysis. This is cleavage of a drug molecule by taking up a molecule of water. Ester + H20 Esterase Acid + Alcohol Similarly amides and polypeptides are hydrolyzed by amidase and peptidases. Hydrolysis occurs in the liver, intestines, plasma, and other tissues. Examples are choline esters, procaine, lidocaine, pethidine, oxytocin. d) Cyclization is formation of a ring structure from a straight chain compound, e.g. proguanil. e) Decyclization is opening up of a ring structure of the cyclic molecule, e.g. phenytoin, barbiturates. Phase II – synthetic (conjugation) reactions These involve conjugation of the drug or its phase I metabolite with an endogenous substrate to form a polar highly ionized organic acid, which is easily excreted in urine or bile. Conjugation reactions have high energy requirements. (1) Glucoronide conjugation is the most important synthetic reaction. Compounds with a hydroxyl or carboxylic acid group are easily conjugated with glucuronic acid, which is derived from glucose, e.g. chloramphenicol, aspirin, morphine, metronidazole, GCS, bilirubin, thyroxine. Drug glucuronides, excreted in bile, can be hydrolyzed in the gut by bacteria, producing beta-glucuronidase. The liberated drug is reabsorbed and undergoes the same fate. This enterohepatic recirculation of some drugs (e.g. chloramphenicol, phenolphthalein, oral contraceptives) prolongs their action. (2) Acetylation. Compounds having amino or hydrazine residues are conjugated with the help of acetyl CoA, e.g. sulfonamides, isoniazid. Multiple genes control the acetyl transferases and rate of acetylation shows genetic polymorphism (slow and fast acetylators). (3) Sulfate conjugation. The phenolic compounds and steroids are sulfated by sulfokinases, e.g. chloramphenicol, adrenal, and sex steroids. The two phases of drug metabolism Synthetic (conjugation) reactions: (4) Methylation. The amines and phenols can be methylated. Methionine and cysteine act as methyl donors. Examples: adrenaline, histamine, nicotinic acid. (5) Ribonucleoside/nucleotide synthesis is important for the activation of many purine and pyrimidine antimetabolites used in cancer chemotherapy, e.g. Xeloda®. (6) Only a few drugs are metabolized by enzymes of intermediary metabolism. Examples: •alcohol by dehydrogenases •allopurinol by xanthine oxidase •succinylcholine and procaine by plasma cholinesterase •adrenaline by monoamine oxidase (MAO) FIRST PASS (PRESYSTEMIC) METABOLISM This refers to metabolism of a drug during its passage from the site of absorption into systemic circulation. All orally administered drugs are exposed to drug metabolism in the intestinal wall and liver in different extent. •High first pass metabolism: propranolol, verapamil, pethidine, salbutamol, nitroglycerine, morphine, lidocaine. •Oral dose of these drugs is higher than sublingual or parenteral dose. •There is individual variation in the oral dose due to differences in the extent of first pass metabolism. •Oral bioavailability is increased in patients with severe liver disease. IV. EXCRETION Excretion is the passage out of systematically absorbed drugs. Drugs and their metabolites are excreted in: urine (through the kidney) •bile and faeces •exhaled air •saliva and sweat •milk •skin The kidney is responsible for excreting all water soluble substances. Glomerular filtration. Glomerular capillaries have large pores. All nonprotein bound drugs (lipid soluble or insoluble) presented to the glomerulus are filtrated. Glomerular filtration of drugs depends on their plasma protein binding and renal blood flow. Glomerular filtration rate (g.f.r.) declines progressively after the age of 50 and is low in renal failure. Tubular reabsorption. Lipid soluble drugs filtrated at the glomerulus back diffuse in the tubules because 99% of glomerular filtrate is reabsorbed, but nonlipid soluble and highly ionized drugs are unable to do so. Thus, the rate of excretion of such drugs, e.g. aminoglycoside (amikacin, gentamicin, tobramycin) parallels g.f.r. Changes in urinary pH affect tubular reabsorption of partially ionized drugs: •Weak bases ionize more and are less reabsorbed in acidic urine. •Weak acids ionize more and are less reabsorbed in alkaline urine. This principle is utilized for facilitating elimination of drugs in poisoning: •Urine is acidified in morphine and atropine poisoning. •Urine is alkalized in barbiturate and salicylate poisoning. The effect of changes in urinary pH on drug excretion is greatest for a drug having pK values between 5 to 8, because only in this case pH dependent passive reabsorption is significant. Tubular secretion is the active transfer of organic acid and bases by two separate nonspecific mechanisms, which operate in the proximal tubules: •Organic acid transport for penicillins, probenecid, salicylates, uric acid, sulfinpyrazones, nitrofurantoin, methotrexate, drug glucuronides, etc. •Organic base transport for thiazides, quinine, procainamide, cimetidine, amiloride, etc. Many drug interactions occur due to competition for tubular excretion, e.g.: •Aspirin blocks uricosuric action of probenecid and sulfinpyrazone and decreases tubular excretion of methotrexate. •Probenecide decreases the urine concentration of nitrofurantoin, increases the duration of penicillin action and impairs excretion of methotrexate. •Quinidine decreases renal and biliary clearance of digoxin by inhibiting efflux carrier P-gp. Tubular transport mechanisms are not well developed at birth. Duration of action of many drugs (penicillins, cephalospoins, aspirin, etc.) is longer in neonates. These systems mature during infancy. •aminoglycosides •beta-lactams •sulfonamides •quinolones •nitrofurans •polymyxins •macrolides •lincosamines •rifampicin •tetracyclines (p.o.) •General inhalation anaesthetics •Potassium iodide •Bronchoantiseptic oils •Alcohol •sulfonamides •barbiturates •reserpine •alcohol •Coffeinum (Caffeine) Rauwolfia serpentina (Reserpine: India) Saliva excretion •oleandomycin •spiramycin •phenytoin •zalcitabine •verapamil Morphine (10% stomach excretion) •morphine pKb: 7.9 •stomach pH: 1–2 •plasma pH: 7.36 Poppy KINETICS OF ELIMINATION (elimination = metabolism + excretion) Clearance (Cl) of a drug is the theoretical volume of plasma from which the drug is completely removed per unit time: Cl = Rate of elimination/Plasma concentration Renal (Clr) or creatinine clearance (Clcr): Curine x Vurine Clrenal = -------------------Cplasma First order (exponential) kinetics. For majority of drugs the processes involved in elimination are not saturated over the clinically obtained concentrations. These drugs have first order kinetics. Their rate of elimination is directly proportional to plasma drug concentration and their clearance remains constant. Semilog plasma concentration-time plot of a drug eliminated by first order kinetics after i.v. injection. Zero order (linear) kinetics. In a few cases where the drugs are inactivated by metabolic degradation (such as ethanol, phenytoin, theophylline, salicylates, and warfarin), the time-course of disappearance of the drug from the plasma does not follow the exponential or biexponential pattern, but is initially linear. These drugs are removed at a constant rate which is independent of plasma concentration. This is often called zero order kinetics. The blood alcohol concentration falls linearly and the rate of fall does not vary with dose. Plasma half live (t1/2) is the time in which the plasma concentration of a drug declines by one half. Drug with long t1/2 can accumulate. Plasma t1/2 of some drugs: Adenosine < 2 sec Dobutamine – 2 min Benzylpenicillin – 30 min Amoxicillin – 1 h Paracetamol – 2 h Atenolol – 7 h Diazepam – 40 h Ethosuccimide – 54 h Digitoxin – 168 h From the peak plasma concentration the drug is virtually eliminated from the plasma in 5 t1/2 periods: (1) (2) (3) (4) (5)