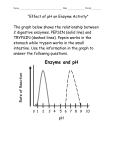
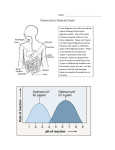
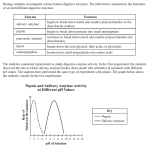
Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Catalytic triad wikipedia , lookup
Peptide synthesis wikipedia , lookup
Genetic code wikipedia , lookup
Enzyme inhibitor wikipedia , lookup
Amino acid synthesis wikipedia , lookup
Biosynthesis wikipedia , lookup
Ribosomally synthesized and post-translationally modified peptides wikipedia , lookup
Biochemistry wikipedia , lookup
Metalloprotein wikipedia , lookup
Discovery and development of neuraminidase inhibitors wikipedia , lookup
Protein Engineering vol.14 no.9 pp.669–674, 2001 N-terminal portion acts as an initiator of the inactivation of pepsin at neutral pH Takuji Tanaka and Rickey Y.Yada1 Department of Food Science, University of Guelph, Guelph, Ontario N1G 2W1, Canada 1To whom correspondence should be addressed. E-mail: [email protected] Porcine pepsin, an aspartic protease, is unstable at neutral pHs where it rapidly loses activity, however, its zymogen, pepsinogen, is stable at neutral pHs. The difference between the two is the presence of the prosegment in pepsinogen. In this study, possible factors responsible for instability were investigated and included: (i) the distribution of positively charged residues on the surface, (ii) an insertion of a peptide in the C-terminal domain and (iii) the dissociation of the N-terminal fragment of pepsin. Mutations to change the number and the distribution of positive charges on the surface had a minor effect on stability. No effect on stability was observed for the deletion of a peptide from the C-terminal domain. However, mutations on the Nterminal fragment had a major impact on stability. At pH 7.0, the N-fragment mutant was inactivated 5.8 times slower than the wild-type. The introduction of a disulfide bond between the N-terminal fragment and the enzyme body prevented the enzyme from denaturing. The above results showed that the inactivation of pepsin was initiated by the dissociation of the N-fragment and that the sequence of this portion was a major determinant for enzyme stability. Through this study, we have created porcine pepsin with increased pH stability at neutral pHs. Keywords: enzyme stability/N-terminal fragment/pepsin/pepsinogen/pH Introduction Pepsin (EC 3.4.23.1) belongs to the aspartic protease family and functions under highly acidic conditions. Enzymes in the aspartic protease family are believed to be ubiquitous and share various similarities in such properties as conformations (Wlodawer et al., 1985; Danley et al., 1989; McKeever et al., 1989; Gilliland et al., 1990; Jaskólski et al., 1990; Newman et al., 1991, 1993; Chen et al., 1992; Tong, et al. 1993; Yang et al., 1997; Lee et al., 1998) and catalytic mechanism (Khan and James, 1998; Richter et al., 1998). Despite the similarities which define the aspartic proteinase family, differences in functionality, e.g. substrate specificities, do exist; thus the aspartic proteinases are ideal models to demonstrate structure– function relationships of enzymes. All proteins are synthesized in a neutral pH environment, thus their natural conformational state and functionality exists in this environment. However, most aspartic proteinases are stable under acidic conditions and become irreversibly denatured under neutral pH conditions (Bohak, 1969; Fruton, 1971). In the case of pepsin, its zymogen, pepsinogen, is stable under neutral pH conditions. Since the majority of the pepsin © Oxford University Press molecule is identical to pepsinogen, minor differences may be responsible for pepsin’s instability at neutral pHs. The most obvious differences between pepsin and pepsinogen are found in the prosegment portion. Not only does the prosegment cover the active site cleft, but it contains a large number of positively charged residues, i.e. 13 (nine Lys, two Arg, two His) out of the 44 residues in the prosegment of pepsinogen are positively charged (James and Sielecki, 1986; Lin et al., 1989). Pepsin on the other hand contains only four positive residues (two Arg, one Lys, one His). Therefore, it is evident that the number and distribution of these positively charged residues contribute to the difference in pH stability between pepsin and pepsinogen. In contrast, unlike most aspartic proteinases, chicken pepsin is relatively stable at neutral pHs (Bohak, 1969). A homology search between porcine and chicken pepsin shows a 74% similarity. Major differences between chicken and porcine pepsins are in charge distribution and the N-terminal amino acid sequences. In chicken pepsin, there are 21 Asp, 13 Glu, 9 Lys, 5 Arg and 4 His charged residues, while in porcine pepsin, 28, 13, 1, 2 and 1 are charged, respectively. For both porcine and chicken pepsin, most of these charged residues are distributed on the surface; however, the distribution of the positively charged residues is more favourable in chicken pepsin. Another difference between pepsin and pepsinogen is the position of the N-terminal fragment. In a zymogenic form and during activation, the N-terminal fragment is in the active site cleft. After activation, this N-terminal fragment is placed in the β-sheet at the bottom of the protein. This relocation results in about a 40 Å movement (James and Sielecki, 1986). Since this portion relocates from one side of the protein to other side, it is suggested that this fragment could be readily moved from its position and could be important for the stabilization of pepsin under neutral pH conditions. Lin et al. reported denaturation of pepsin begins with the denaturation of the Nterminal domain, and suggested that the interaction between the N-fragment and the enzyme body is important for stability (Lin et al., 1993). A recent crystallographic study of cathepsin D, another neutral pH stable aspartic proteinase, showed that the N-terminal fragment of this enzyme is relocated to its active site cleft at pH 7.5, resembling its zymogenic form (Lee et al., 1998). From the above observations, several key factors in the instability of pepsin are suggested. In this study, the following were undertaken in order to investigate and possibly improve the stability of pepsin at neutral pHs: (i) the alteration of the charge distribution on the surface of pepsin, (ii) the deletion of the extra amino acid sequence observed in porcine pepsin C-domain, (iii) the addition of hydrogen bond stabilizers and (iv) the stabilization of the N-terminal portion of pepsin through mutations and the introduction of a disulfide bond. Materials and methods Materials The expression plasmid pTFP1000, which contains the gene for wild-type porcine pepsinogen fused to the 3⬘ end of the 669 T.Tanaka and R.Y.Yada gene for thioredoxin, was constructed according to Tanaka and Yada (Tanaka and Yada, 1996). Restriction endonuclease NheI and DNA-modifying enzymes were purchased from Life Technologies (Gaithersburg, MD). Restriction endonucleases XbaI and BglII were from Boehringer Mannheim (Laval, PQ, Canada) and New England Biolabs (Mississauga, ON, Canada), respectively. Oligonucleotide primers used for mutagenesis and the synthetic octapeptide KPAEFF(NO2)AL (ss 1) were synthesized at the Institute of Molecular Biology and Biotechnology at McMaster University (Hamilton, ON, Canada). The synthetic oligopeptide LSF(NO2)NleAL methyl ester (ss 2) was obtained from Sigma Chemical (St Louis, MO). Casamino acids were purchased from Difco (Detroit, MI). DEAE Sepharose CL-6B was from Pharmacia Biotech (Uppsala, Sweden). All other reagents were of the purest grade commercially available. Mutagenesis of pepsinogen Site-directed mutagenesis was carried out by the method of Zoller and Smith (Zoller and Smith, 1982) using a uracilcontaining single-stranded M13mp19 DNA template (Kunkel, 1985). The following oligonucleotide primers were used: G2C: 5⬘-CCG CTG CCC TCA TAT GCG ATG AGC C-3⬘; G2S/D3Y: 5⬘-GCCCTGATATCATATGAGCCC-3⬘; L10M/T12A/E13S: 5⬘-GAACTACATGGATGCATCGTACTTTGGC-3⬘; S46K: 5⬘-GTCTACTGCAAAAGCTTAGCCTGCAGC-3⬘; D52N/N54K/Q55R: 5⬘-CCTGCAGCAACCACAAGCGCTTCAACCC-3⬘; D60K: 5⬘-CAACCCTGATAAATCGTCGACCTTCGAG-3⬘; L167C: 5⬘-CGATGCCGCCGCACAGTACTACACTGCCGC-3⬘; S196R: 5⬘-CAGATTACCCTCGATCGCATCACC-3⬘; D200G/E202K: 5⬘-CACCATGGCCGGCAAGACCATCG-3⬘; ∆240–246/⫹GD: 5⬘-GGAGCCAGCGAGGGCGATATCAGCTGCTCC-3⬘; S254K: 5⬘-CTGCTCCTCAATTGACAAGCTGCCTGAC-3⬘. Some mutations were also incorporated with other mutations. The following combinations of mutations were constructed: G2C/L167C; G2S/D3Y [N-frag(A)]; G2S/D3Y/L10M/T12A/E13S (N-frag); L10M/T12A/E13S [N-frag(B)]; S46K/D52N/N54K/Q55R/D60K (N-DOM); S196R/D200G/E202K (C-DOM); S46K/D52N/N54K/Q55R/D60K/S196R/D200G/E202K (N⫹C); ∆240–246/⫹GD (Del). G2C/L167C, N-frag(A), N-frag and N-frag(B) were intended to stabilize the N-terminal portion of pepsin. N-DOM, C-DOM and N⫹C were made to alter the charge distribution. Del was undertaken to remove the putative mobile portion in the C-terminal domain. Expression of Trx-PG Mutant pepsinogen DNA was cloned into the expression plasmid pTFP1000, which contains the gene for wild-type pepsinogen, by replacing the corresponding fragment of mutant pepsinogen DNA using restriction enzymes. The resultant plasmids were used to transform Escherichia coli GI724 by the method of Hanahan (Hanahan, 1983). Escherichia coli GI724 cells carrying the plasmid were cultured in 1.5 l of induction media (1⫻ M9 salts, 0.2% casamino acids, 0.5% glucose, 1 mM magnesium 670 chloride) containing 0.15 mg/ml ampicillin at 30°C. When the culture reached early log phase (absorbance at 550 nm ⫽ 0.5), expression of Trx-PG was induced through the addition of 15 ml of a 10 mg/ml L-tryptophan solution. The culture was incubated for a further 6 h, after which the cells were harvested by centrifugation at 8000 g for 10 min at 4°C. Isolation and purification of Trx-PG Expressed Trx-PG was extracted from the cells by sonication and purified through ammonium sulfate precipitation followed by chromatography on anion-exchange columns DEAE-Sephacel and DEAE-Sepharose CL-6B. Purified Trx-PG was dialysed against 20 mM Tris–HCl buffer pH 7.5, for 2 h at 4°C, filter sterilized and stored at 4°C. Purity was assessed by SDS–PAGE, according to Laemmli (Laemmli, 1970), followed by staining with Coomassie Blue. Kinetics Preparation of pepsin for each mutant Trx-PG and determination of the kinetic parameters were as previously described (Tanaka and Yada, 1996). Pepsin was obtained from Trx-PG by acidification and purification through gel filtration. Kinetics were measured using peptide ss 1 and ss 2 at a substrate concentration range of 0.01–0.2 mM at 37°C. pH employed were pH 2.1 for ss 1 and pH 3.95 for ss 2. The protein concentration was determined as the amount of pepstatin required to completely inhibit pepsin. Stability tests Recombinant pepsinogen was activated and the corresponding pepsin was purified through Sephadex G-50 gel filtration with 20 mM sodium acetate buffer (pH 5.3). An aliquot of 40 µl of this preparation (0.1 mg/ml protein) was mixed with 40 µl of dH2O and 80 µl of 0.4 M buffer (sodium phosphate buffer pH 7.0, 7.5, 8.0). Small aliquots were withdrawn at specific times and quenched with 3 M sodium acetate buffer (pH 5.2). Residual activity was determined with the synthetic peptide substrate (ss 1) at pH 2.1, 25°C. To investigate the effects of additives, the same stability test was conducted; however, a 40% (v/v) glycerol/40% (w/v) sucrose solution was used to replace the dH2O in the above test. Disulfide bonds do not spontaneously form under pH 6.0, thus to accelerate the formation of the disulfide bond in the G2C/L167C mutant, several oxidizing reagents were tested. The pepsin preparation was incubated in 0.1 M Tris–malate buffer (pH 4.0, 5.0, 6.0, 6.6, 7.2) with an oxidizing reagent, i.e. 5 mM K3Fe(CN)6, 5 mM FeCl3, 1 mM o-iodothobenzoate, 0.5 mM dithionitrobenzoate or 0.5 mM dithiopyridine, for 24 h at room temperature (or 4°C). After oxidization, each sample was tested for stability. Results and discussion Kinetics of activated pepsin No mutations studied in this work were made at or near the active site. It was anticipated that the mutations conducted in this study would not affect enzyme activity although our previous study on the sole lysine residue on pepsin showed a single mutation outside of the active site could change enzyme activity (Cottrell et al., 1995). Reaction kinetics were, therefore, determined for two synthetic substrates, KPAEFF(NO2)AL and LSF(NO2)NleAL (Table I). Near the optimum pH of pepsin (pH 2.1), mutants had k0/Km values of 0.52–2.88 while wild-type had a k0/Km value of 1.92. The activities of mutants were very high, thus the structures were likely to be affected at small N-terminal fragment and neutral pH inactivation of pepsin Table I. Kinetic constants of mutant and wild-type pepsin Enzyme ss 1 (KPAEFF(NO2)AL) Km (µM) Wild-type N⫹C DEL N-frag N-frag(A) N-frag(B) G2C/L167C 34 57 70 83 75 65 80 ⫾ ⫾ ⫾ ⫾ ⫾ ⫾ ⫾ 5 5 6 15 8 9 7 ss 2 (LSF(NO2)NleAL methyl ester) k0 (/s) k0/Km (/s/µM) Km (µM) 65.4 ⫾ 3.1 77.5 ⫾ 2.9 60.9 ⫾ 2.6 68.9 ⫾ 6.6 186.0 ⫾ 9.8 187.1 ⫾ 12.8 41.6 ⫾ 2.0 1.92 ⫾ 0.30 1.36 ⫾ 0.13 0.87 ⫾ 0.08 0.83 ⫾ 0.17 2.48 ⫾ 0.30 2.88 ⫾ 0.44 0.52 ⫾ 0.03 19 31 15 28 28 41 18 ⫾ ⫾ ⫾ ⫾ ⫾ ⫾ ⫾ 6 13 2 7 7 5 1 k0 (/s) k0/Km (/s/µM) 190.5 ⫾ 18.9 50.7 ⫾ 9.4 25.7 ⫾ 1.0 80.7 ⫾ 10.2 122.0 ⫾ 15.6 315.3 ⫾ 16.8 13.0 ⫾ 0.22 10.0 1.64 1.71 2.88 4.36 7.69 0.72 ⫾ 3.3 ⫾ 0.75 ⫾ 0.24 ⫾ 0.81 ⫾ 1.22 ⫾1.02 ⫾ 0.17 Values represents the mean of three determinations ⫾ SD. Reactions were carried out at pH 2.1 for ss 1 and pH 3.95 for ss 2, both at 37°C. Table II. Rate constants of inactivation of mutants at pH 7.0 Enzyme kd (/min) N-frag N-frag (glycerol, sucrose) N-frag (pH 7.5) G2S/D3Y [N-frag(A)] L10M/T12A/E13S [N-frag(B)] G2C/L167C N⫹C N-DOM C-DOM DEL Wild-type Wild-type (glycerol) Wild-type (sucrose) Wild-type (glycerol, sucrose) Wild-type (pH 7.5) 0.0169 ⫾ 0.0014 0.00229 ⫾ 0.00031 0.268 ⫾ 0.008 0.0680 ⫾ 0.0038 0.0646 ⫾ 0.0015 0.0536 ⫾ 0.0051 0.0421 ⫾ 0.0071 0.0905 ⫾ 0.0173 0.0852 ⫾ 0.0237 0.0851 ⫾ 0.0088 0.0991 ⫾ 0.0075 0.0365 ⫾ 0.0026 0.0203 ⫾ 0.0009 0.00743 ⫾ 0.00216 ND (too rapid) ND, inactivation was too rapid (completed within 5 min) under the experimental conditions to determine the inactivation rate. Values represent the means of three determinations ⫾ SD. degrees. At a higher pH (pH 3.95), all the mutants showed lower activities, although the activities were high if their low Km values are considered. Stability and additives The time course of inactivation of wild-type and mutant pepsin is shown in Figure 1. The inactivation rate constants were calculated from the plots and summarized in Table II. The inactivation of wild-type pepsin was measured at pH 7.0 and 7.5. At pH 7.0, pepsin was rapidly inactivated at a rate of 0.099/min (open circles in Figure 1A). At this rate, virtually all activity ceased within 1 h. At pH 7.5, pepsin was rapidly inactivated and lost activity in 5 min (open circles in Figure 2B). Lin et al. suggested that inactivation was caused by irreversible denaturation which was triggered by the denaturation of the N-terminal domain (Lin et al., 1993). Therefore, it may be possible to stabilize the protein by preventing this denaturation. In a previous study, it was found that glycerol was able to preserve unstable mutants of fused pepsinogen (Richter et al., 1999). Thus a hydroxyl group rich compound may aid in reducing denaturation. In the present study, glycerol and sucrose, another hydroxyl group rich compound, were chosen. Both additives had no effect on the activity of wild-type under the experimental conditions employed. At 20% (w/v), both sucrose and glycerol slowed inactivation (Figure 1A). The rate of inactivation (Table II) was reduced to 0.0203 and 0.0365/min at pH 7.0, respectively. Ten to 30% of the initial activity was retained after 60 min at pH 7.0 using Fig. 1. Inactivation of wild-type and mutant pepsin. Pepsin was inactivated in the conditions described in the text and residual activities were plotted. (A) Open circle, wild-type; closed square, wild-type with glycerol; open triangle, wild-type with glycerol and sucrose. (B) Closed circle, wild-type; open triangle, N⫹C; closed triangle, N-fragment; open circle, ∆240–246; open square, N-frag(A); closed square, N-frag(B). Each data point represents the mean of three determinations. these additives. Moreover, the combination of 20% glycerol and sucrose reduced the inactivation rate to 0.00743/min and resulted in half of the initial activity after 60 min. Denaturation can result from either the rearrangement of the hydrogen bonds and/or electrostatic interactions. Since denaturation is triggered by a rise in pH, electrostatic interactions are most likely to be involved. Although electrostatic interactions are not affected by hydroxyl groups, the addition of glycerol/ sucrose did help stabilize pepsin. At the same time, the addition of glycerol/sucrose did not result in 100% stabilization since some pepsin activity was still lost; therefore, both electrostatic interactions and hydrogen bonds are believed to contribute to the denaturation of pepsin. Stability of the mutants (charge distributions) Change of the distribution of negatively charged residues on the surface Pepsin has only a few positively charged residues but an abundance of negatively charged residues on its surface. Given that pepsin is an acidic protein, it is reasonable to expect that 671 T.Tanaka and R.Y.Yada Fig. 3. Comparison of the sequence of the N-terminal fragment. From the comparison of porcine and chicken pepsin sequences, three mutants were planned. Chicken pepsin has three extra amino acids on the N-terminal end. The addition of these amino acids to porcine was not considered because it could interfere with the activation itself. The substitution amino acid residues in the mutants are indicated using underlining. Fig. 2. Inactivation of the N-terminal fragment mutant. Inactivation test of N-fragment, N-frag(A) and N-frag(B) mutants showed that individual mutations in the N-fragment mutant were not critical for the stabilization of pepsin. Both N-frag (A) and (B) mutants showed slight stabilization effects (B) and were similar in stability (A) to the wild-type while the N-frag mutant showed drastic stabilization. (A) The inactivation reactions were carried out at pH 7.0. (B) The inactivation reactions were carried out at pH 7.5. Symbols are: wild-type (open circle); N-frag(A) (inverted open triangle); N-frag(B) (open square); N-fragment (upright open triangle); wild-type with glycerol and sucrose (closed circle); N-frag(A) with glycerol and sucrose (inverted closed triangle); N-frag(B) with glycerol and sucrose (closed square); N-fragment with glycerol and sucrose (upright closed triangle). Each data point represents the mean of three determinations. many negatively charged residues occur on the surface. As pH increases, these residues become deprotonated resulting in electrostatic repulsion which may lead to instability. Thus replacement of these negatively charged residues with neutral or positive residues may stabilize pepsin at neutral pH values. The results of the mutation Replacements of the positive residues were chosen in the region where negatively charged residues were highly concentrated. Mutations were selected in order to resemble the sequence of chicken pepsin. Kinetic measurements at pH 2.1 and 3.95 were taken for the resultant mutant, N⫹C. The mutant showed comparable kinetic constants to the wild-type; therefore, mutations on the surface had little effect on the kinetic parameters (Table I). Although the N⫹C mutant had similar activity as wild-type, the stability at neutral pH changed (Table II; Figure 1B). The mutant inactivated at about half the rate of the wild-type. The inactivation of the mutant, which had mutations on only the Nor C-terminal domains (N-DOM and C-DOM, respectively), did not show the same level of stabilization. The rate of the inactivation of N-DOM and C-DOM was comparable to that of the wild-type. The results of the above three mutants demonstrated that the distribution of the negatively charged residues on the surface of the porcine pepsin helped to stabilize the enzyme, however, the degree of stabilization was not substantial. Since Lin et al. had shown that the initial denaturation occurred in the N-terminal domain (Lin et al., 1993), the mutations on the N-terminal domain were expected to have a dominant effect. 672 The negatively charged residues on the N-terminal domain themselves, however, only had a minor effect on the stability. Thus the distribution of the charges over the entire surface must be changed in order to achieve noticeable stabilization of pepsin at neutral pH. Stability of the mutants (N-terminal fragment) Possible effect of the N-terminal portion on denaturation The position of the N-terminal fragment in pepsin changes from the active site cleft to the opposite side of the molecule during activation (James and Sielecki, 1986). This implies that the Nterminal portion is relatively easily displaced from its original position. If this N-terminal portion moves away from its original position, a β-sheet loses one of its strands, raising the possibility of destabilization of the sheet and/or interaction with another hydrogen bond source. Therefore, it is suggested that stabilizing this fragment in its original position, which is a strand of a βsheet on the bottom of pepsin molecule, will prevent denaturation in the event of neutralization. In order to examine this possibility, mutations were introduced into the N-terminal portion to keep it in the fixed position at the bottom of the pepsin molecule. The comparison of the amino acid sequence of the pepsins from porcine and chicken (chicken pepsin is relatively stable at neutral conditions) revealed major differences in the N-terminal portion. We chose five amino acid residues to be replaced (Figure 3). Lin et al. suggested that Asp11 in the N-terminal portion was critical to the denaturation at neutral pH (Lin et al., 1993). However, the sequence comparison of pig and chicken pepsins showed that this position in chicken pepsin was conserved, thus this residue was not chosen as a mutation site. The results of the mutation The mutation of the N-terminal portion was done with two sets of mutations, G2S/D3Y [N-frag(A)] and L10M/T12A/E13S [N-frag(B)]. These two mutants and a third mutant which combined both mutations (N-frag) exhibited similar kinetic constants to the wild-type and had comparable catalytic activities for both synthetic substrates (Table I). Stability tests of the N-frag mutant showed that it was stabilized by 5.8 times as compared to the wild-type (Table II; Figure 2A). While the wild-type was inactivated in 60 min, this mutant retained 30% of its activity after 60 min. Even after 4 h, 1.8% of the original activity was observed with this mutant. When the pH was raised to 7.5 (Figure 2B), the wild-type showed no activity after 5 min. Even the addition of glycerol and sucrose, which was shown to stabilize the enzyme, had no effect. When the N-frag mutant was exposed to pH 7.5, it retained 5% activity after 15 min. The rate constant of inactivation for N-frag at pH 7.5 was 0.268/min. Moreover, in the presence of both glycerol and sucrose at pH 7.5, the enzyme retained 25% activity after 30 min. N-terminal fragment and neutral pH inactivation of pepsin Fig. 4. Molecular model comparison of wild-type and N-terminal fragment mutant pepsin. Superposition of the N-terminal fragment of wild-type (grey) and the N-frag mutant (black) models. There were no major differences. Root mean square deviation between backbone atoms of wild-type and N-frag was 0.28 Å. The numbers show the mutation sites in N-frag. Since the N-frag mutant had five amino acid replacements, some of the mutations could be more critical than others. The G2S/D3Y and L10M/T12A/E13S mutants, however, showed less stability than the N-frag mutant (Figure 2). Either mutant showed an inactivation rate which was about 1.5 times slower than the wild-type while the N-frag mutant was 5.8 times slower. Also, at pH 7.5, the activities of both N-frag(A) and N-frag(B) mutants were quenched slower than the wild-type, but faster than the N-frag mutant. After 5 min, N-frag(A), N-frag(B), Nfrag and wild-type had 0, 3, 27 and 0% of the original activity in the absence of glycerol and sucrose while 45, 46, 72 and 0% of the original activities remained in the presence of glycerol and sucrose, respectively. Molecular model minimization showed that these mutations did not contribute to the internal interactions (Figure 4). The only major difference between the wild-type and N-frag was the addition of a hydrogen bond between Ser2 γ-oxygen and L167 nitrogen in the G2S mutation. However, kinetics of the N-frag(A) mutant showed this addition was insufficient to stabilize the protein entirely. Thus it was concluded that each of the five replacements by themselves were not critical, but synergistically helped to stabilize the enzyme. Since each mutation in itself was not critical to stabilization, the mechanism of stability was still in question. The authors suggest two possibilities on how the mutations stabilized pepsin: (i) the release of the N-terminus portion is prevented with these mutations or (ii) these mutation sites are responsible for the stability of the released N-terminal portion. The crystal structure of inactive cathepsin D at pH 7.5 showed that the N-terminal portion is relocated into the active site cleft and is stabilized by an interaction to the catalytic site (Lee et al., 1998). From this crystal structure data, the authors suspect the second possibility is more likely. Disulfide bond formation to fix the N-terminal portion in place If the mobility of the N-terminal portion initiates denaturation, then it would be logical to think that fixing this portion to the enzyme body would prevent denaturation. To fix the N-terminus portion to the enzyme body, a potential disulfide bond was introduced. This mutant, G2C/L167C, had a cysteine residue at the second residue of the N-terminal portion and another cysteine on the opposite side of the enzyme body (Figure 5). The kinetic studies of this mutant showed lower, but a substantial amount of activity as compared to the wild-type (Table I). The rate constant of the inactivation of G2C/L167C at pH 7.0 was about half of the wild-type but was not as slow as N-frag. However, inactivation slowed down after 30 min, and Fig. 5. Molecular model of the introduced disulfide bond in G2C/L167C mutant pepsin. A molecular model around the mutation site of G2C/L167C is shown. The introduced disulfide bond, shown by the arrow, is well accommodated in this position. Fig. 6. Inactivation of G2C/L167C mutant pepsin. Inactivation of G2C/L167C mutant and the effect of the oxidizing reagent are shown. At pH 7.5, the oxidization did not stabilize the wild-type [without oxidization (open square) and with oxidization (closed square)] while oxidization of G2C/L167C stabilized the enzyme [without oxidization (open circle) and with oxidization (closed circle)]. Each data point represents the mean of three determinations. had a low but noticeable activity (3.2%) over 24 h while the N-frag, N-frag(A) and N-frag(B) mutants were completely inactive after 24 h. These results would imply that the formation of the disulfide bond prevented denaturation by preventing the movement of the N-terminal portion. However, the slower disulfide bond formation seemed to compete with the faster inactivation since substantial activity was lost before reaching the plateau. To increase the rate of disulfide bond formation, oxidizing reagents were used, i.e. FeCl3, K3Fe(CN)6, o-iodothobenzoate, dithionitrobenzoate and 3,3⬘-dithiopyridine. These reagents had comparable effects. After 24 h of oxidization with 2 mM FeCl3, the oxidized G2C/L167C was tested for stability at pH 7.5 (Figure 6). G2C/L167C, without oxidizers, showed slower inactivation than wild-type; however, most of the activity was lost after 480 min. In the presence of FeCl3, inactivation slowed down and reached a plateau at 20% of its initial activity. Further inactivation studies showed that activities were retained, 11% at 24 h and 5% at 74 h. These results indicated that the disulfide bond formation kept the N-terminal fragment close to its native position, thereby, stabilizing the enzyme. The formation of disulfide bonds was faster at pH 7.5 than at pH 7.0, thus a greater stabilizing effect was observed at the higher pH. Conclusion The present study began with the question of which factors destabilize pepsin at neutral pH. From the differences observed 673 T.Tanaka and R.Y.Yada among aspartic proteases and between zymogens and activated enzymes, the authors speculated that both the distribution of the charges on the surface and the stability of the N-terminal portion on the enzyme body were vital for stability. The former possibility was investigated by introducing a number of positive charges in place of negative charges. The results indicated that these positive charges resulted in a minor improvement in stability. Therefore, it is suggested that the distribution of the charges had an effect only after the denaturation was triggered by other factors. The mutations on the N-terminal portion exhibited a larger contribution to restricting denaturation than the mutations to alter charge distribution. Replacement of five amino acid residues in the first 13 residues reduced the inactivation rate by 5.8 times at pH 7.0. Moreover, in the presence of glycerol and sucrose, this mutant showed a very low rate of inactivation, 0.00229/ min, and the residual activity after 240 min was 50% of the initial activity compared to the wild-type which lost most of its activity in 60 min. This mutation indicated the importance of the N-terminus to denaturation. Subsequently, the introduction of a disulfide bond was used to fix the N-terminal portion onto the enzyme body. Since inactivation was faster than disulfide bond formation, a large amount of the activity was initially lost, after which, the rate of inactivation slowed and then plateaued. These results demonstrated that limiting the N-terminal mobility limits the denaturation of pepsin at neutral pH. From the above results, the following was concluded: (i) the distribution of the charges was important in the stability of pepsin, (ii) the N-terminal portion was a key segment to inactivation and (iii) the N-terminal portion was the trigger to denaturation of the N-terminal domain causing inactivation. Acknowledgements The authors are grateful to Ms Erika Tsumura for her technical assistance, Mr Kenneth Payie and Mark Yoshimasu for their helpful suggestions on preparation of the manuscript. This research was supported by a grant from the Natural Sciences and Engineering Research Council of Canada. References Bohak,Z. (1969) J. Biol. Chem., 244, 4638–4648. Chen,L., Erickson,J.W., Rydel,T.J., Park,C.H., Neidhart,D., Luly,J. and AbadZapatero,C. (1992) Acta Crystallogr. B, 48, 476–488. Cottrell,T.J., Harris,L.J., Tanaka,T. and Yada,R.Y. (1995) J. Biol. Chem., 271, 19974–19978. Danley,D.E., Geoghgan,K.F., Scheld,K.G., Lee,S.E., Merson,J.R., Hawrylik,S.J., Rickett,G.A., Ammirati,M.J. and Hobart,P.M. (1989) Biochem. Biophys. Res. Commun., 165, 1043–1050. Fruton,J.S. (1971) In Boyer,P.D. (ed.), The Enzyme. Academic Press, NY, pp. 119–164. Gilliland,G.L., Winborne,E.L., Nachman,J. and Wlodawer,A. (1990) Protein, 8, 82–101. Hanahan,D. (1983) J. Mol. Biol., 166, 557–580. James,M.N.G. and Sielecki,A.R. (1986) Nature, 319, 33–38. Jaskólski,M., Miller,M., Rao,J.K.M., Leis,J. and Wlodawer,A. (1990) Biochemistry, 29, 5889–5898. Khan,A.R. and James,M.N.G. (1998) Protein Sci., 7, 815–836. Kunkel,T.A. (1985) Proc. Natl Acad. Sci. USA, 82, 488–492. Laemmli,U.K. (1970) Nature, 227, 680–685. Lee,A.Y., Gulnik,S.V. and Erickson,J.W. (1998) Nat. Struct. Biol., 5, 866–871. Lin,X.-l., Loy,J.A., Sussman,F. and Tang,J. (1993) Protein Sci., 2, 1383–1390. Lin,X.-l., Wong,R.N.S. and Tang,J. (1989) J. Biol. Chem., 264, 4482–4489. McKeever,B.M., Navia,M.A., Fitzgerald,P.M., Springer,J.P., Leu,C.-T., Heimbach,J.C., Herber,W.K., Sigal,I.S. and Darke,P.L. (1989) J. Biol. Chem., 264, 1919–1921. Newman,M., Safro,M., Frazao,C., Khan,G., Zdanow,A., Tickle,I.J., Blundell,T.L. and Andreeeva,N. (1991) J. Mol. Biol., 221, 1295–1309. Newman,M., Watson,F., Roychowdhury,P., Jones,H., Badasso,M., Cleasby,A., Wood,S.P., Tickle,I.J. and Blundell,T.L. (1993) J. Mol. Biol., 230, 260–283. 674 Richter,C., Tanaka,T., Koseki,T. and Yada,R.Y. (1999) Eur. J. Biol., 261, 746–752. Richter,C., Tanaka,T. and Yada,R.Y. (1998) Biochem. J., 335, 481–490. Tanaka,T. and Yada,R.Y. (1996) Biochem. J., 315, 443–446. Tong,L., Pav,S., Pargellis,C., Dô,F., Lamarre,D. and Anderson,P. (1993) Proc. Natl Acad. Sci. USA, 90, 8387–8391. Wlodawer,A., Miller Maria Jaskólski,M., Sathyanarayana,B.K., Baldwin,E., Weber,I.T., Selk,L.M., Clawson,L., Schneider,J. and Kent,S.B.H. (1985) Science, 245, 616–621. Yang,J., Teplyakov,A. and Quail,J.W. (1997) J. Mol. Biol., 268, 449–459. Zoller,M.J. and Smith,M. (1982) Nucleic Acids Res., 10, 6487–6500. Received February 26, 2001; revised May 9, 2001; accepted June 15, 2001